
LIMS for Contract Research Organizations: A Complete Guide to GCP Compliance and Multi-Sponsor Data Management
Audit failure cost
4-9×
Cost of a repeated study from audit trail failure vs. preventing the gap at data entry
FDA warning letters
72%
Of FDA warning letters to CROs cite data integrity failures as a primary violation
GCP standard update
ICH E6(R3)
Revised GCP guideline mandating risk-based quality management - effective 2025
Contract research organizations carry a structurally unique operational burden - running parallel studies for multiple sponsors on shared laboratory infrastructure, while keeping every dataset perfectly isolated, every audit trail fully defensible, and every protocol deviation documented before it becomes a regulatory finding. This guide examines what a purpose-configured CRO LIMS must do, where generic laboratory software consistently falls short, and how to evaluate whether a system is genuinely ready for a regulated multi-sponsor environment.
Key takeaways
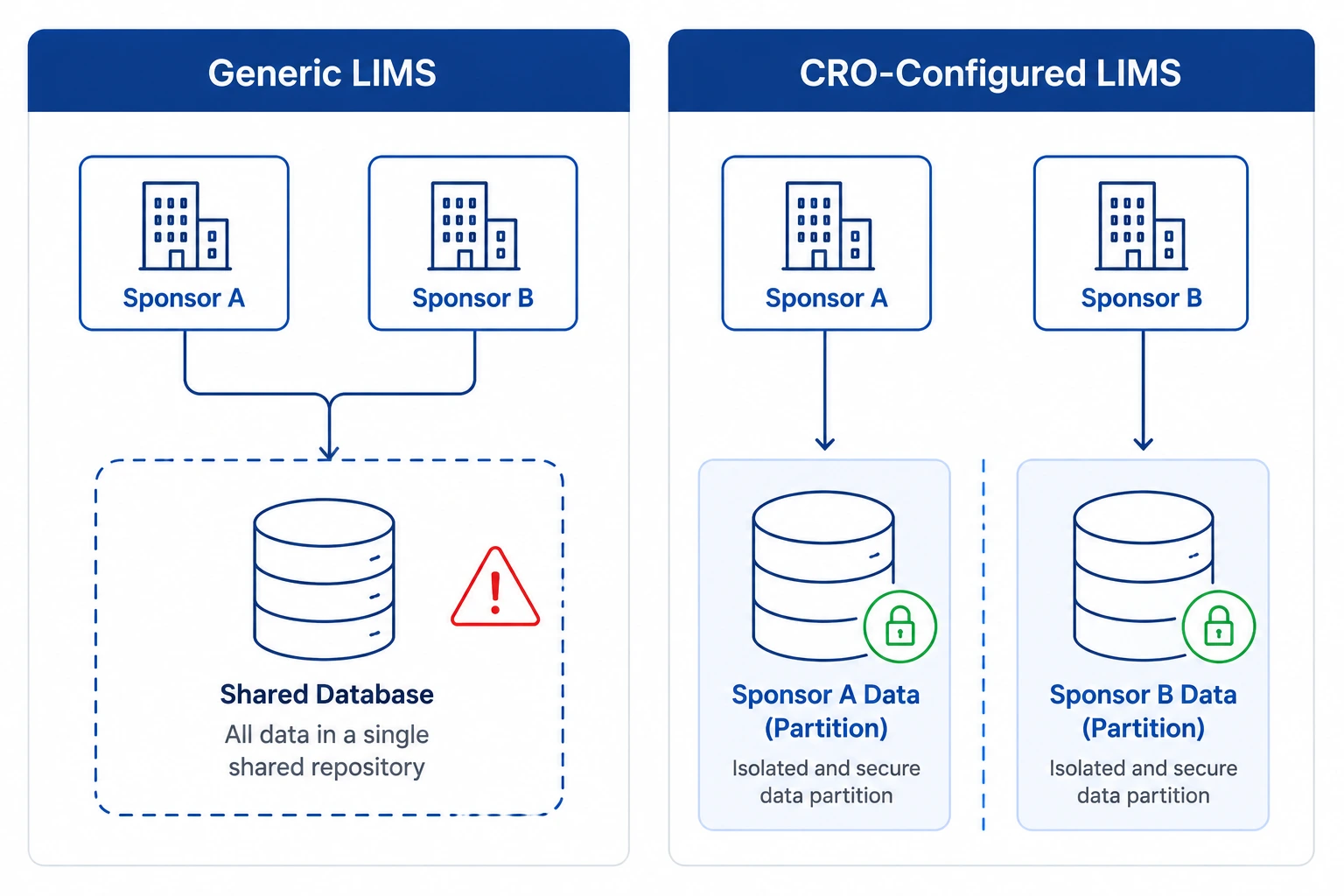
- Multi-sponsor CRO operations require database-level data segregation - naming conventions and manual folder structures are not sufficient for 21 CFR Part 11 or ICH E6(R3) compliance.
- A purpose-configured CRO LIMS enforces protocol parameters at the point of activity - deviations are prevented prospectively, not discovered retrospectively during data review.
- Key compliance frameworks for CRO labs include ICH E6(R3) GCP, 21 CFR Part 11, ISO/IEC 17025, and FDA 21 CFR Part 58 (GLP) - often simultaneously on the same infrastructure.
- OOS investigation workflows must be managed inside the LIMS with mandatory Phase I and II investigation records, CAPA linkage, and closure verification.
- Sponsor data packages assembled manually from multiple systems introduce transcription errors and cannot meet regulatory timeline demands - automated package generation is required.
What this article covers
- Why CRO lab management is structurally different
- The 6 critical challenges a CRO LIMS must solve
- GCP, 21 CFR Part 11, ISO/IEC 17025, and GLP - compliance by framework
- Generic vs. Revol LIMS 8.0 - a direct comparison
- What to look for when evaluating a CRO LIMS vendor
- Frequently asked questions
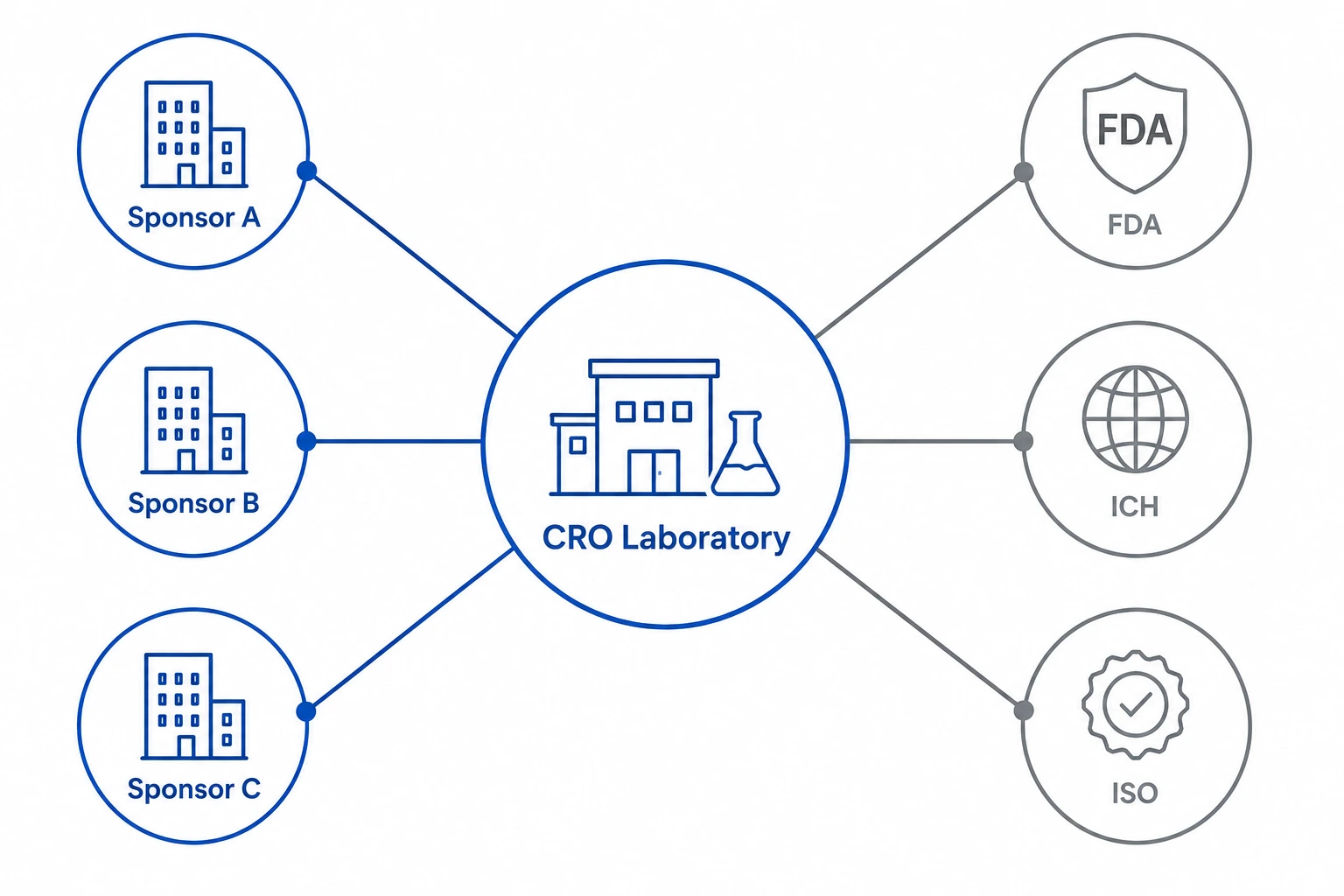
1. Why CRO lab management is structurally different
A contract research organization is not simply a laboratory that runs clinical trials. It is a multi-client service organization that must maintain the scientific and regulatory integrity of several independent studies simultaneously - often in the same physical space, using the same instruments, performed by the same personnel, under different sponsor SOPs, different protocol requirements, and different regulatory jurisdictions.
This structural reality creates a category of data management problem that standard laboratory software was never designed to handle. When a general-purpose LIMS is deployed in a CRO environment, it typically addresses sample tracking and results storage adequately. What it does not address is the layer of controls that makes multi-sponsor CRO operations defensible: sponsor-level data segregation, protocol-specific workflow enforcement, multi-site chain of custody management, and audit trail architecture satisfying both 21 CFR Part 11 and ICH E6(R3).
The consequences of these gaps are not theoretical. A shared instrument log that commingles data from two sponsor studies creates an audit finding that neither sponsor will accept. A missing analyst signature on a deviation report triggers a query that can stall a clinical timeline by weeks. A result released without a second-reviewer electronic sign-off fails 21 CFR Part 11 even if the underlying analytical work was performed correctly.
A LIMS configured for CRO operations must address the full complexity of the environment - not just the analytical workflow, but the contractual, regulatory, and multi-sponsor data architecture that makes every result defensible to every client and every regulatory authority simultaneously.
2. The 6 critical challenges a CRO LIMS must solve
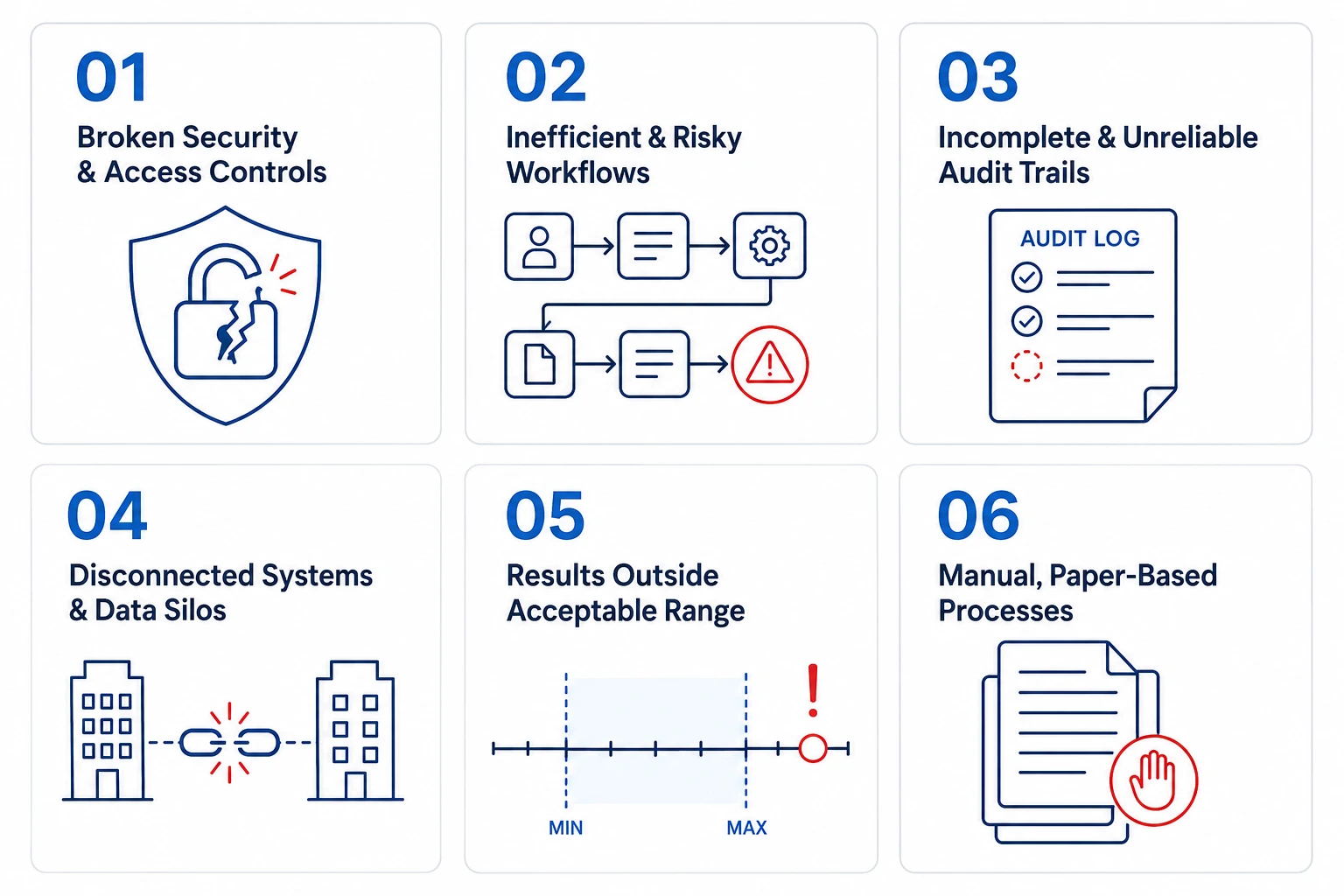
In most CROs running a generic LIMS, sponsor isolation relies entirely on user discipline: naming conventions, manual folder structures, and the assumption that analysts will not access a study they are not assigned to. When instrument data is ingested from a shared LC-MS or plate reader, raw files enter a common repository. There is no system-level barrier between Sponsor A's Phase II oncology samples and Sponsor B's Phase I toxicology panel running on the same instrument the same afternoon. Cross-contamination of records — even read-only access — is a 21 CFR Part 11 violation and a GCP non-conformance under ICH E6(R3) Section 5.5.
- Study-level data partitioning enforced at the database architecture level - no analyst can query, view, or modify records outside their assigned sponsor study without an explicit, logged role assignment.
- Instrument data ingestion mapped to study IDs at the point of import - raw files automatically tagged to the originating protocol, preventing orphaned data entries shared across studies.
- Cross-study access attempts logged as security events, generating automatic alerts to the quality manager without manual monitoring.
- Sponsor-specific result dashboards and report templates ensure deliverable packages contain only the authorised study's data - no manual filtering required.
Each sponsor delivers a study protocol with specific sample handling requirements: defined centrifugation parameters, maximum processing delay windows, required aliquot volumes, storage temperature specifications, and analytical sequence logic. In a generic LIMS, these are documented in an attached protocol PDF that analysts are expected to consult before each activity. The system records what was done - not whether what was done was correct for that study. Protocol deviations are discovered retrospectively during data review, not prevented at the point where they occur.
- Protocol parameters imported at study setup and enforced as system-level workflow logic - the system prevents progression to the next step if current parameters fall outside protocol-defined ranges.
- Processing delay timers triggered automatically at sample receipt - alerts fire before the protocol window closes, not after it has been missed.
- Required fields and mandatory checkpoints defined per sponsor study - a deviation from expected parameters forces a documented deviation record before the workflow can continue.
- Protocol version control built in - if a sponsor issues an amendment mid-study, the system tracks which samples were processed under which SOP version across the amendment boundary.
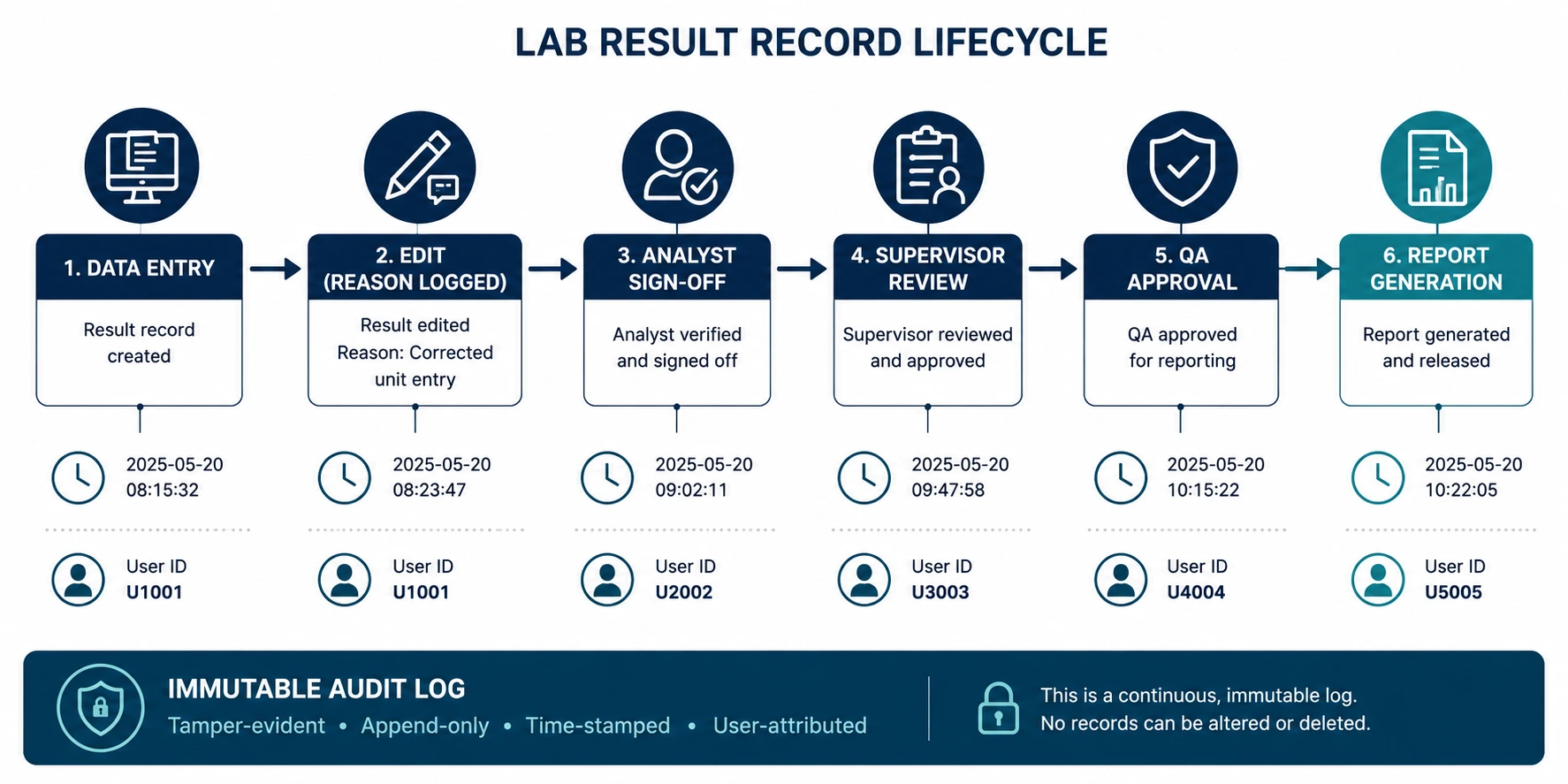
21 CFR Part 11 requires that every record alteration - including deletions, overwrites, and access events where data was subsequently used - be captured with a time-stamped, operator-identified audit entry that cannot be modified. In practice, many CROs operate systems that log result entries but not result edits; that capture analyst actions but not reviewer access events; and that allow administrators to suppress log entries without a separate audit record of the suppression. During an FDA inspection, these gaps are not minor procedural findings - they are data integrity violations that can invalidate an entire study dataset.
- Immutable audit trail captures every record creation, modification, deletion, and access event - including who viewed the record, when, and from which workstation.
- Reason-for-change mandatory on every edit - the original value, new value, rationale, and the editor's authenticated identity are all preserved in the same audit record.
- Audit log access itself is logged - even an administrator reviewing the audit trail generates an entry, preventing silent administrative access to regulated records.
- 21 CFR Part 11-compliant electronic signatures applied at defined workflow checkpoints: analyst release, supervisor review, QA approval, and sponsor-facing report authorisation.
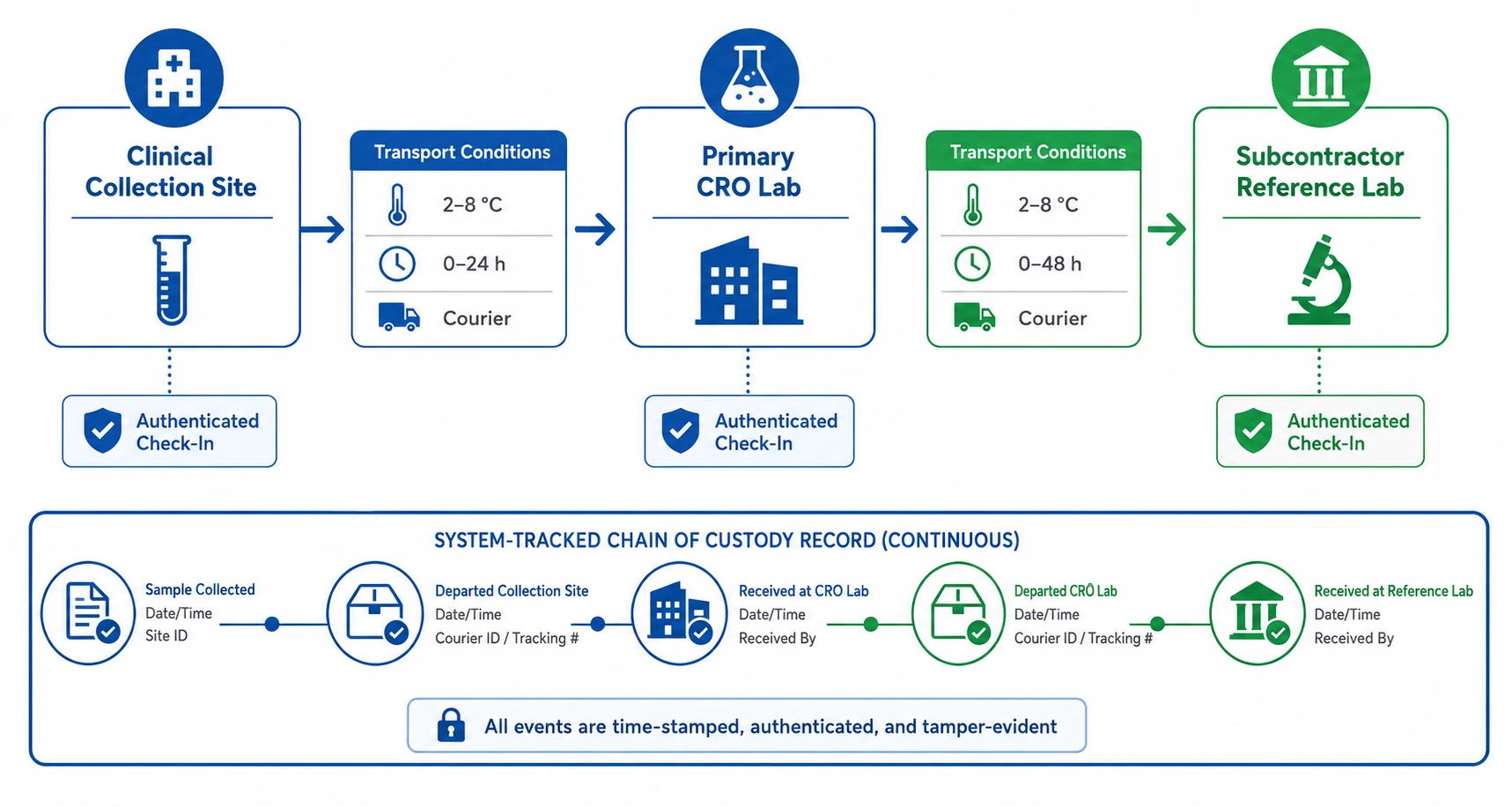
CROs frequently subcontract specialist assays to reference laboratories or operate across multiple sites. When samples leave the primary facility, the chain of custody typically transfers to a paper manifest, a courier tracking number, and an email confirmation from the receiving site. There is no system-level record that samples arrived in the expected condition, that transport temperature requirements were met, or that samples were received by an authorised individual. Under ICH E6(R3) and FDA GCP, the CRO retains responsibility for all subcontracted work - and that responsibility cannot be discharged with a tracking number.
- Inter-site transfer workflows managed within the system: dispatch records include sample condition at shipment, required transport conditions, authorised receiving personnel, and expected receipt window.
- Receipt confirmation at the destination site requires an authenticated system check-in - discrepancies between dispatched and received condition trigger a formal deviation record.
- Subcontractor performance tracked across studies - systematic receipt delays, condition deviations, or turnaround failures surfaced in quality dashboards, supporting vendor qualification.
- Complete chain of custody document generated on demand for any sample, covering the full journey from clinical collection through primary processing through subcontracted analysis.
When an analytical result falls outside specification, the investigation typically begins in a spreadsheet or a separate quality management system, proceeds through email chains, and concludes with a verbal approval that may or may not generate a written record. The system records the original OOS result and the final retest — but the investigation, root cause, corrective action, and authorisation to release or reject the sample exist outside the auditable record. An OOS event with a missing investigation record is treated in an FDA inspection as an uninvestigated OOS — one of the most serious findings a regulated laboratory can receive.
- OOS results trigger automatic investigation workflows within the system at the point of data entry - the result cannot be released or retested until the investigation is formally initiated.
- Phase I investigation (laboratory error assessment) and Phase II investigation (root cause analysis) managed as structured workflows with mandatory fields and defined timelines.
- CAPA records linked directly to the originating OOS result - every corrective action has an owner, a deadline, and a verification step before the CAPA can be closed.
- OOS history analysed across studies in quality dashboards - recurring failure modes identified before they become systematic regulatory findings.
At the end of each study phase, the CRO must deliver to the sponsor a validated data package: raw results, audit trails, deviation reports, QC summary, instrument calibration records, and certificate of analysis — correctly attributed, formatted to sponsor requirements, and free of data from any other study. In most CROs, this package is assembled manually from multiple systems. The process takes days, introduces transcription errors, and must be partially rebuilt from scratch whenever the sponsor requests changes.
- Sponsor-configured report templates store the exact output format each client requires - results, metadata, audit trail excerpts, and QC summaries assembled automatically from system records.
- Data package generation is a single authorised system action - the system pulls only records associated with the sponsor's study, applies their template, and produces the complete deliverable without manual extraction.
- The data package carries a system-generated integrity checksum - the sponsor can verify that the delivered data was not modified after generation.
- Interim reporting uses the same workflow - sponsors requesting mid-study snapshots receive consistently formatted, audit-backed reports without additional analyst effort.
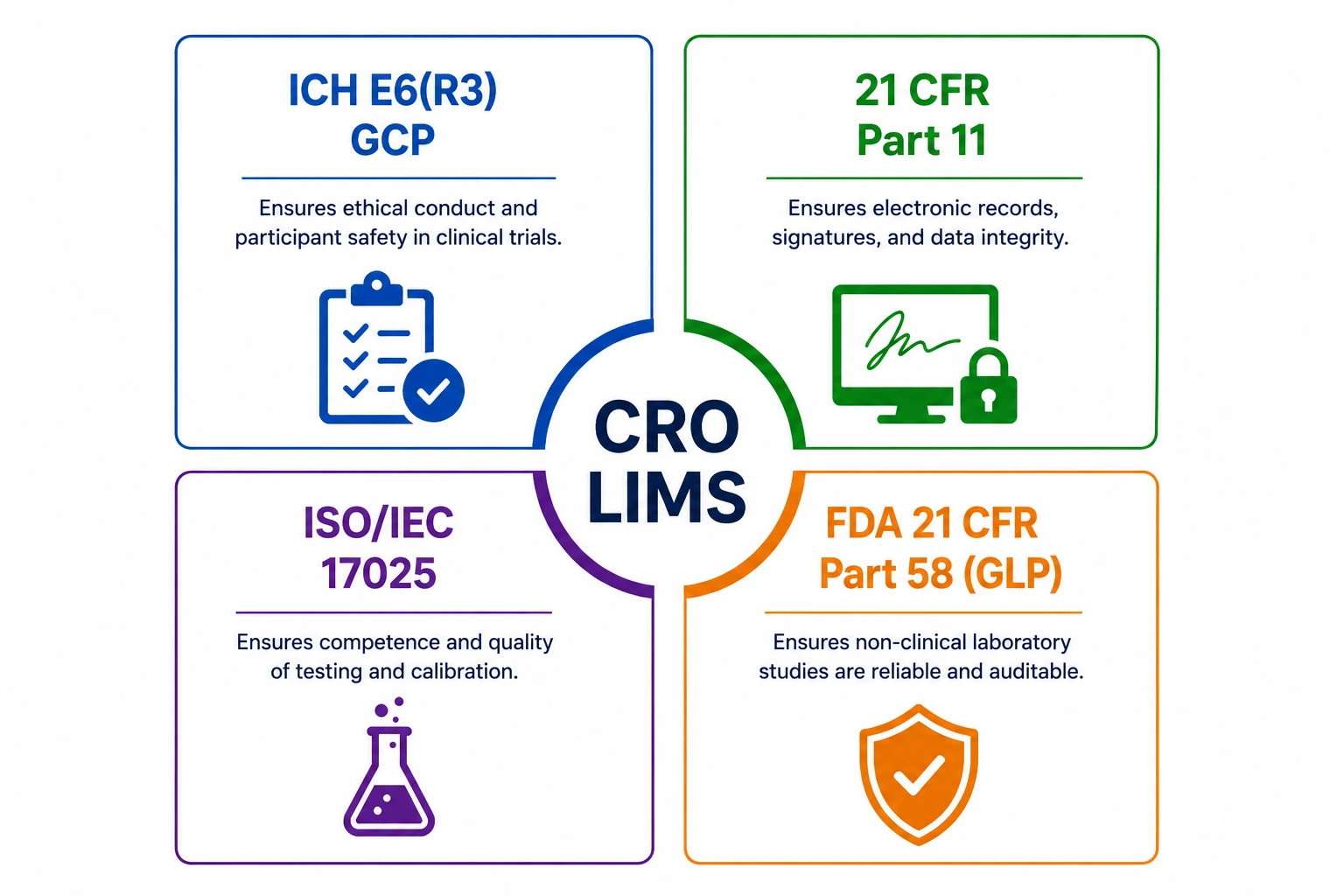
3. GCP, 21 CFR Part 11, ISO/IEC 17025, and GLP - compliance by framework
CROs typically operate under multiple simultaneous regulatory frameworks - ICH E6(R3), 21 CFR Part 11, ISO/IEC 17025, and GLP - because different sponsors, different trial phases, and different study types demand different compliance architectures. A well-architected CRO LIMS, including Revol LIMS 8.0, sets the compliance configuration at the study level during protocol setup — the system then enforces only the requirements applicable to that study's regulatory context. A GLP toxicology study and a GCP Phase II trial can run within the same instance, each governed by its own framework, with no risk of applying the wrong compliance logic to the wrong study type.
4. Generic vs. Purpose Built CRO LIMS - a direct comparison

5. What to look for when evaluating a CRO LIMS vendor
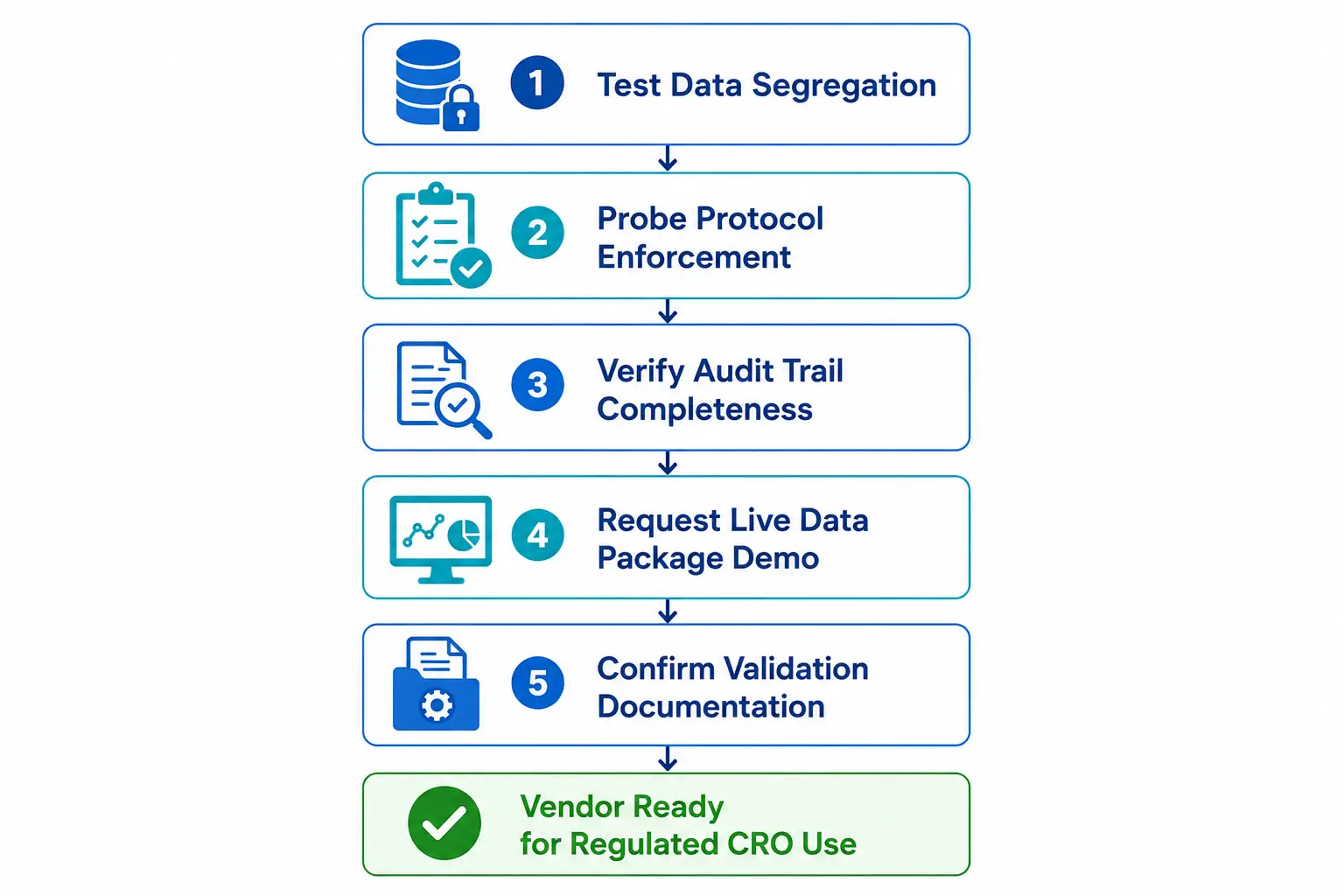
The five evaluation prompts below are designed to surface real capability gaps - not feature lists. Use them in vendor demonstrations before any procurement decision.
Test the data segregation model with a live scenario
Ask the vendor to demonstrate what happens when an analyst assigned only to Study A attempts to access a result record from Study B. The system should structurally prevent the access - not generate a dismissible warning. If the only barrier is a user-level permission an administrator can grant without a formal access request record, the segregation model is not robust enough for a regulated multi-sponsor environment.
Probe the protocol enforcement architecture
Ask the vendor to simulate a processing delay exceedance - a sample received at 08:00 but not centrifuged until 14:00 against a four-hour protocol window. The system should have alerted the analyst before the window closed and forced a documented deviation record before the workflow could proceed. If it recorded timestamps but allowed the workflow to continue uninterrupted, it is tracking compliance retrospectively, not enforcing it prospectively.
Verify 21 CFR Part 11 audit trail completeness
Request a complete audit trail for a single result record - from initial data entry through any edits, through result review and QA sign-off, through report generation. Every record should be timestamped, operator-identified, and reason-for-change annotated. Then ask the vendor to show the audit trail entry generated when an administrator viewed the audit trail itself. If that event is not logged, the trail has a structural gap a Part 11 inspection will identify.
Request a live sponsor data package generation
Ask for a live demonstration of generating a complete data package for a demo study - not a pre-built PDF, but an on-demand generation from system records in real time. If the process takes more than a few minutes and requires vendor implementation support, the reporting architecture is not production-ready for a CRO generating packages across multiple concurrent studies on regulatory timelines.
Confirm validation documentation scope
Ask for the IQ/OQ/PQ validation documentation package for CRO-specific workflows. It should include test scripts specific to GCP protocol enforcement, 21 CFR Part 11 audit trail completeness, GLP study director approval chains, and multi-sponsor data segregation. A vendor offering only generic laboratory validation scripts has not validated the CRO-specific configuration - site-specific PQ testing will need to cover the gap before use in a regulated study.
Have questions about CRO LIMS implementation for your lab?
Contact Our Team →6. Frequently asked questions
The following questions reflect real queries from CRO lab directors and quality managers evaluating LIMS for regulated multi-sponsor environments.
Q1. Can a pharma-configured LIMS be adapted for CRO multi-sponsor operations?
A pharma LIMS is typically configured for a single sponsor's SOPs applied consistently across all studies. Adapting it for multi-sponsor CRO use requires building study-level partitioning, configurable protocol enforcement, and sponsor-specific reporting templates that most pharma implementations do not include. The most important due diligence question to ask any vendor is: how many multi-sponsor CROs are currently running this system in a 21 CFR Part 11-validated state, and can you provide a reference contact from one of them?
Q2. What is the difference between audit trail tracking and audit trail enforcement?
Tracking records what happened - it logs operator actions and timestamps them. Enforcement prevents non-compliant actions from occurring — it makes it structurally impossible to edit a released result without a reason-for-change entry, or to release a sample with an open OOS investigation. ICH E6(R3) and 21 CFR Part 11 require both. A system that tracks comprehensively but does not enforce workflow integrity at the point of activity satisfies the documentation requirement but not the data integrity requirement.
Q3. How should a CRO LIMS handle protocol amendments issued mid-study?
Purpose-configured systems manage protocol amendments as version-controlled events. When a sponsor issues an amendment, the new protocol version is imported and activated with a defined effective date. Samples processed before the effective date retain their original protocol version reference in the audit trail; samples processed after it are governed by the amended version. This creates a documentable record of which version applied to which samples — critical when a regulatory authority asks why handling parameters changed mid-study.
Q4. What LIMS validation scope is required for a CRO operating under 21 CFR Part 11?
Validation must cover every regulated workflow: electronic signature chain integrity, audit trail completeness across all record-altering events, access control enforcement at the study and record level, OOS investigation workflow, and data package generation. IQ, OQ, and PQ protocols must use CRO-specific test scripts. Every sponsor-specific configuration that differs from the validated baseline requires a change control record and, where the change affects regulated workflows, supplemental PQ testing before use in a regulated study.
Q5. Can a single CRO LIMS instance support multiple global regulatory jurisdictions simultaneously?
Study-level framework configuration allows a CRO to operate GCP studies under ICH E6(R3) for EU-based sponsors, 21 CFR Part 11 studies for US FDA submissions, and ISO/IEC 17025-accredited reference testing - all within the same LIMS instance. Each study is configured at setup with the applicable regulatory framework, and the system enforces only the requirements relevant to that study's jurisdiction, without requiring separate instances for each jurisdiction served. Revol LIMS 8.0 supports this multi-jurisdiction configuration natively.
Q6. What are the most common reasons CRO LIMS implementations fail?
The most frequent failure patterns involve three avoidable decisions: deploying a standard laboratory LIMS without CRO-specific configuration; underestimating the validation scope required for a multi-framework regulated environment; and failing to involve sponsor QA contacts in acceptance testing - leading to post-go-live discoveries that system outputs do not meet individual sponsor reporting requirements. A successful implementation begins with a detailed workflow mapping exercise documenting every sponsor's specific requirements before configuration begins, and includes sponsor-representative data package review as part of user acceptance testing.
Continue reading
- Biobank LIMS: Solving Sample Management Challenges and ISO 20387 Compliance
- How to Prepare Your LIMS for an FDA Audit: A Practical Checklist
- Achieving ISO/IEC 17025 Compliance: How a Modern LIMS Simplifies Accreditation
- Toxicology Lab Workflows: Why Generic LIMS Implementations Fail and How to Fix Them
Author: Revol LIMS Team · marketing@revollims.com · www.revollims.com





 (1) (1).webp)



.webp)










