
Toxicology Lab Workflows: Why Generic LIMS Implementations Fail and How to Fix Them
A toxicology laboratory is not a clinical lab that happens to test for drugs. It is not a general chemistry lab that happens to use mass spectrometry. It is a distinct operational environment with a specific set of workflows, chain of custody requirements, confirmation testing protocols, cut-off management rules, and medico-legal reporting obligations that have no clean equivalent in most other laboratory disciplines.
That distinction matters enormously when it comes to LIMS implementation — and it is the reason so many toxicology labs end up with systems that technically work but practically fail. The LIMS was built for or configured around a generic laboratory workflow. The toxicology lab's specific needs were approximated, customised around, or ignored during implementation. The result is a system the lab has learned to tolerate rather than one it can actually rely on.
This article examines the seven most common reasons generic LIMS implementations fail in toxicology environments, and what a properly configured system for this discipline needs to include. It is written for laboratory directors, QA managers, and IT decision-makers at clinical toxicology, forensic toxicology, workplace drug testing, and pain management monitoring laboratories.
What This Article Covers
- Why toxicology workflows are fundamentally different from the generic lab environments most LIMS platforms are designed around
- The 7 most common implementation failures in toxicology LIMS deployments — each with a root cause diagnosis and a concrete fix
- A side-by-side comparison of generic vs. toxicology-configured LIMS behaviour across key workflow steps
- What to look for when evaluating or re-configuring a LIMS for a toxicology environment
- FAQs
1. What Makes Toxicology Lab Workflows Different
Before diagnosing why generic LIMS implementations fail, it helps to be precise about what makes toxicology workflows unusual. Most laboratory disciplines are organised around a linear process: receive sample, perform test, report result. Toxicology layered on top of that a set of requirements that introduce significant complexity at every stage.
Chain of Custody Is Not Optional — It Is the Product
In clinical and forensic toxicology, the integrity of the result is inseparable from the documented chain of custody. A positive finding that cannot be traced back to an unbroken, documented chain of sample collection, transfer, accessioning, aliquoting, analysis, and reporting is not a finding the lab can defend. This is especially true in workplace drug testing governed by SAMHSA/DOT guidelines, in medico-legal investigations, and in pain management monitoring where results may be used in clinical or legal proceedings.
A LIMS that tracks samples adequately for a clinical chemistry or food testing lab — logging who received what and when — may be wholly inadequate for toxicology, where every custody transfer must be explicitly documented, every aliquot must be traceable to its parent specimen, and every access to a regulated sample must leave an audit trail.
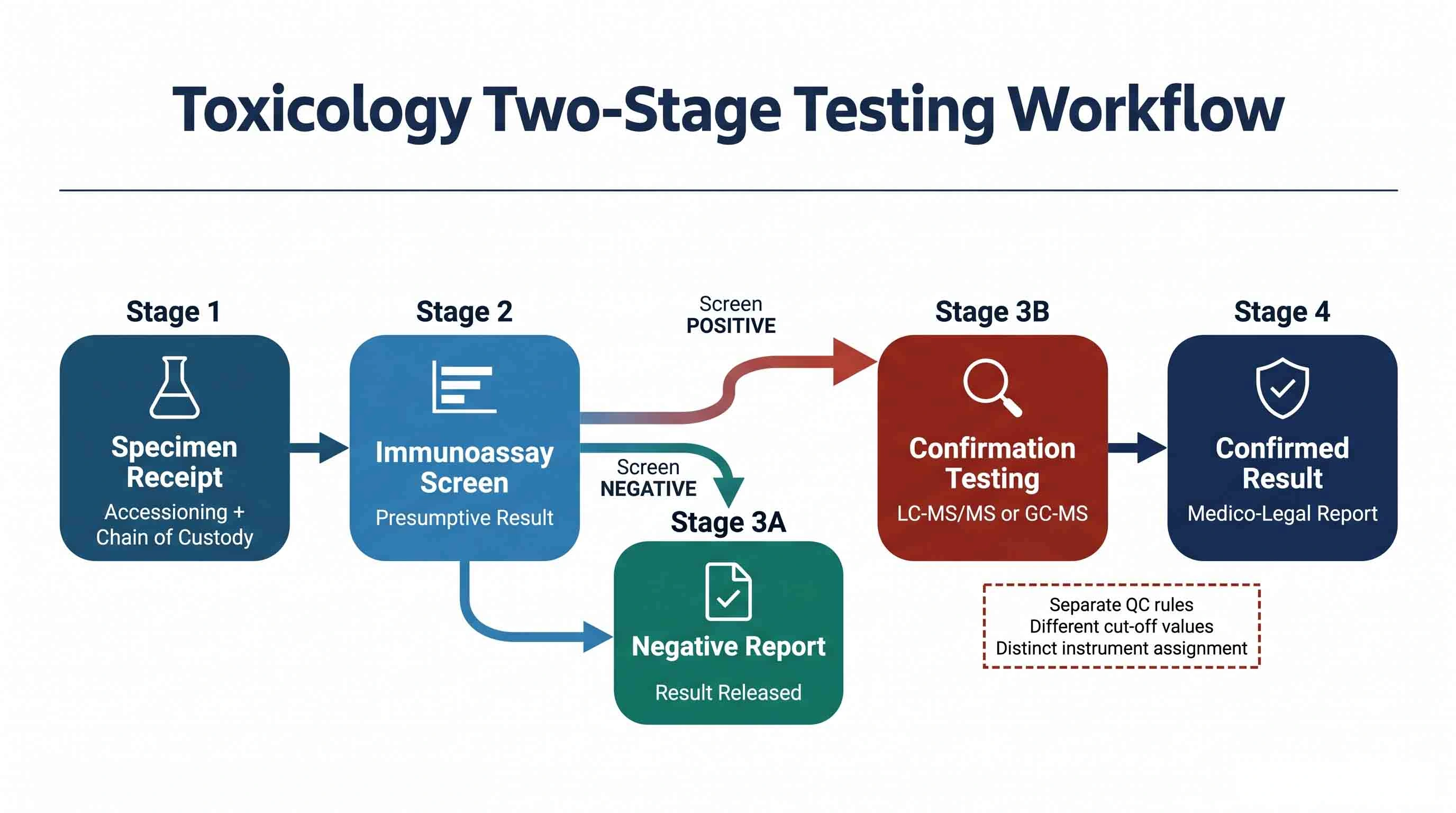
Screening and Confirmation Are Two Distinct Workflows, Not One
Most toxicology testing follows a two-stage structure. An initial immunoassay screen identifies presumptive positives. All presumptive positives then proceed to a confirmatory test — typically LC-MS/MS or GC-MS — which uses a different method, a different cut-off, and produces a result with a completely different evidential weight. These are not two sequential steps in one workflow. They are two separate analytical processes with separate QC requirements, separate instrumentation, and in many contexts, separate reporting.
A generic LIMS that treats a sample's journey as a single linear progression from receipt to result will struggle to model this bifurcation cleanly — particularly when presumptive positive rates drive variable workloads into the confirmation stream, and when the lab needs to track screen-negative, screen-positive-confirmed-negative, and screen-positive-confirmed-positive samples in separate reporting pathways.
Cut-Off Management Is Complex and Context-Dependent
Cut-off concentrations in toxicology are not fixed laboratory parameters. They vary by drug class, by testing context (workplace vs. clinical vs. forensic), by the regulatory framework governing the client (DOT vs. non-DOT for workplace testing, for example), and in some cases by individual client contract. A lab serving multiple client types may need to apply different cut-off values to the same analyte for different specimen batches — and the LIMS must manage this without risk of cross-application.
Instrument Integration Goes Deeper Than Data Transfer
Toxicology labs are among the heaviest users of hyphenated chromatographic techniques — LC-MS/MS, GC-MS, GC-MS/MS. These instruments generate raw data files that are not just results: they include ion ratio data, retention time information, qualifier ion chromatograms, and calibrator/QC performance metrics that are all part of the analytical record. A LIMS that captures the numerical result from these instruments but discards the supporting raw data is not meeting the analytical record retention requirements that forensic and regulated toxicology demands.
2. The 7 Most Common LIMS Implementation Failures in Toxicology Labs

These failures are not theoretical. They are patterns observed across toxicology LIMS implementations at labs of varying sizes and disciplines — clinical, forensic, workplace drug testing, and pain management monitoring. Some are failures of configuration. Some are failures of scope. All are avoidable.
Failure 1 — Chain of Custody Enforcement Is Incomplete or Manual
▸ The Problem
The most common and consequential failure. A generic LIMS may log sample receipt and result entry, but leave the intervening custody transfers — from receiving to accessioning, from accessioning to the screening bench, from the screening bench to confirmation, from confirmation to the MRO reporting queue — undocumented or documented only through manual paper logs that run parallel to the system.
The effect is a LIMS that contains a record of who received a sample and who reported the result, but has no defensible documentation of what happened in between. In a workplace drug testing context governed by 49 CFR Part 40, this gap is a regulatory exposure. In a forensic investigation, it can render a positive result legally indefensible.
▸ Real-World Consequence
A laboratory discovers during an external audit that its LIMS does not capture the transfer of samples from the screening bench to the confirmation analyser. The gap is covered by paper chain of custody forms that are scanned and stored separately. The auditor notes that the electronic record and the paper record have inconsistencies — a finding that triggers a corrective action and a backlog review.
How to Fix It
- The LIMS must enforce custody transfer at every physical handoff point — not just at receipt and reporting. Every transfer should require an authenticated user action that timestamps the event and logs both the relinquishing and receiving parties.
- Aliquot tracking must be bidirectional — every aliquot must be traceable to its parent specimen, and the parent specimen record must show all aliquots derived from it and their current disposition.
- Paper chain of custody forms, where still required by regulation (as in DOT/SAMHSA testing), should be scanned and linked to the specimen record in the LIMS at accessioning, not filed separately.
- Any access to a regulated specimen — including storage retrieval, aliquot preparation, and witnessed destruction — should generate a system record with user ID and timestamp.
Failure 2 — Screening and Confirmation Workflows Are Not Separated

▸ The Problem
Generic LIMS platforms model a sample moving through a single workflow: received, tested, resulted. In toxicology, this model does not fit. Presumptive positive screens need to trigger a separate confirmation workflow — with a different test method, different cut-off values, different instrumentation, different QC requirements, and potentially different turnaround time expectations. This is not just a routing change; it is a fundamentally different analytical and reporting process.
When a LIMS cannot model this distinction cleanly, labs typically manage the bifurcation manually — flagging samples on printed lists, moving them to a separate bench, and entering confirmation results as if they were primary test results. The LIMS record no longer accurately reflects the analytical process, creating documentation gaps and making it difficult to track screen-to-confirmation conversion rates or identify analytical trends.
▸ Real-World Consequence
A clinical toxicology lab cannot produce a reliable report of its immunoassay screen positive rates by analyte, because screen-positive samples that proceed to confirmation are manually removed from the standard workflow and re-entered as separate test requests. The LIMS has no linked record connecting the screen result to the confirmation result for the same specimen.
How to Fix It
- The LIMS workflow must be configured to automatically route presumptive positive specimens to a confirmation worklist without manual intervention.
- Screen result and confirmation result for the same specimen should be linked in the LIMS record — the confirmation result should reference the originating screen result and the same chain of custody.
- Separate QC rules, cut-off parameters, and instrument assignment should be configurable for the confirmation workflow independent of the screening workflow.
- Reporting templates should be able to produce a final result that integrates the screen and confirmation findings — with the confirmation result taking precedence for any specimen that proceeded through both stages.
Failure 3 — Cut-Off Values Are Hardcoded or Managed Outside the LIMS
▸ The Problem
In a generic LIMS configured for clinical chemistry or microbiology, reference ranges and decision thresholds tend to be relatively stable and uniform. In toxicology, cut-off values are variable by design. A lab running DOT-regulated urine drug testing and non-DOT employer testing simultaneously must apply different cut-offs to the same analyte for the same specimens on the same day, depending on which regulatory framework governs each donor.
When a LIMS cannot manage this variability natively, labs resort to maintaining cut-off values in external spreadsheets, manually applying them to batches, or hardcoding a single value and making manual exceptions. Each workaround introduces the risk of cut-off misapplication — a result flagged or cleared incorrectly because the wrong threshold was applied to the wrong client batch.
▸ Real-World Consequence
A workplace drug testing laboratory receives a complaint from a client whose non-DOT samples were processed using DOT cut-off values. Investigation reveals that the LIMS only supports a single global cut-off per analyte. The lab had been managing client-specific cut-offs via a spreadsheet that an analyst updates manually at the start of each batch. On the day in question, the spreadsheet was not updated before processing began.
How to Fix It
- The LIMS must support client-level or order-level cut-off configuration — the ability to assign different threshold values for the same analyte depending on the test ordering context.
- Cut-off values should be version-controlled and audit-trailed — any change to a cut-off parameter should be logged with who changed it, when, and what the previous value was.
- The system should prevent result release for any specimen where the applicable cut-off value has not been positively confirmed at the batch or order level.
- For multi-panel testing, the LIMS should manage cut-offs per analyte per panel per client — this is a non-trivial configuration requirement that should be explicitly scoped during any LIMS evaluation.
Failure 4 — LC-MS/MS and GC-MS Integration Captures Results But Not Raw Data
▸ The Problem
Toxicology's confirmatory testing relies heavily on chromatographic mass spectrometry — techniques that produce not just a numerical concentration result but a body of supporting raw data: ion chromatograms, ion ratios, retention times, internal standard performance, and calibration curve data. In a forensic or legally regulated context, this raw data is part of the analytical record and must be retained and retrievable.
Many LIMS integrations with LC-MS/MS and GC-MS systems are configured to capture only the final quantitative result — the concentration value that appears on the report. The raw instrument output file, the ion ratio data that would allow an independent expert to verify the identification, and the system suitability records that demonstrate the instrument was performing within specification are either not captured or stored in a separate instrument-specific system with no link to the LIMS specimen record.
▸ Real-World Consequence
During a legal challenge to a positive forensic toxicology result, the lab is asked to produce the complete analytical record for the specimen including raw instrument data. The LIMS record shows the final concentration. The raw LC-MS/MS data file exists on the instrument workstation but has no direct link to the LIMS record and cannot be conclusively tied to the specific analytical run without manual reconstruction.
How to Fix It
- The LIMS integration with chromatographic instruments should capture or link to the full raw data file — not just the extracted result. At minimum, the LIMS record should contain a direct file path reference or embedded metadata that uniquely identifies the instrument run file.
- Ion ratio data, internal standard recovery, and system suitability results should be imported and stored as part of the analytical record for each specimen, not just the quantitative result.
- Calibration and QC data for each analytical run should be linked to all specimens processed in that run, so that run-level QC failures can be applied retrospectively to affected specimens.
- Instrument raw data retention should be addressed in the LIMS data retention policy — the system should enforce retention periods that meet the laboratory's longest applicable regulatory requirement.
Failure 5 — QC and Calibration Rules Are Generic, Not Analyte-Specific
▸ The Problem
Analytical quality control in toxicology is not a single set of rules applied uniformly across all testing. Different assay types, different matrices (urine, blood, oral fluid, hair, meconium), and different regulatory frameworks carry different QC expectations. Immunoassay QC rules differ from mass spectrometry QC rules. DOT-regulated testing has specific QC requirements for calibrators and controls that differ from clinical toxicology practice.
When a generic LIMS applies a single QC schema across all test types, the result is either over-restriction — failing batches that would be acceptable under the applicable method-specific criteria — or under-restriction — releasing batches that contain QC failures not caught by a schema designed for a different method. Neither outcome is acceptable in a regulated or medico-legal context.
▸ Real-World Consequence
A toxicology lab runs urine immunoassay screening and confirmatory LC-MS/MS from the same LIMS. The QC configuration was set up using the same rules for both. When a calibration failure specific to the mass spectrometry run occurs, the LIMS does not flag it because the QC rule was designed around the immunoassay platform's acceptance criteria. The batch is released. The failure is identified during an internal QC review two days later.
How to Fix It
- QC rules must be configurable at the method and instrument level, not applied globally. The LIMS should support separate QC schemas for immunoassay screening, GC-MS confirmation, LC-MS/MS confirmation, and any other analytical platform in use.
- Calibration curve acceptance criteria — r² values, back-calculation accuracy, bias limits — should be configurable per analyte panel and should prevent result release for any specimen in a batch where calibration criteria are not met.
- For DOT/SAMHSA-regulated testing, the LIMS should enforce the specific QC requirements of 49 CFR Part 40, including the required positive, negative, and blank controls at specified frequencies.
- Failed QC events should trigger automatic batch hold — the system should not allow result release for any specimen in an affected batch without explicit QA override with documented justification.
Failure 6 — Specimen Validity Testing Is Not Integrated Into the Workflow
▸ The Problem
Urine specimen validity testing (SVT) — creatinine, specific gravity, pH, oxidant testing — is a mandatory component of DOT-regulated workplace drug testing and a standard practice in many clinical toxicology contexts. The SVT result is not supplementary information; it determines the reportable status of the specimen. A specimen flagged as substituted or adulterated is reported as such, not as a drug test result, and the reporting pathway and the regulatory consequences are entirely different.
A generic LIMS that does not have a native SVT workflow may handle validity testing as a separate test order, with results that have no automatic connection to the drug test disposition. The lab manually cross-references the two sets of results to determine the specimen's reportable status. This manual step is a source of documentation gaps and reporting errors — especially at volume.
▸ Real-World Consequence
A high-throughput workplace drug testing lab processes several hundred specimens per day. Specimen validity testing is performed on a separate analyser and results are entered into the LIMS as a separate test order. On one occasion, a substituted specimen — where creatinine is below the DOT threshold for substitution — is reported as a negative drug result because the analyst processing the drug test did not receive the SVT flag. The error is caught by the MRO before report release, but the near-miss triggers a corrective action.
How to Fix It
- Specimen validity testing must be integrated into the main specimen workflow, not treated as a separate test order. The LIMS should automatically flag any specimen where SVT results trigger a regulatory threshold — substituted, adulterated, or invalid — and prevent drug test result release until the SVT disposition is confirmed.
- For DOT/SAMHSA testing, the specific SVT thresholds for substitution (creatinine <2 mg/dL or specific gravity <1.0010 or >1.0200) and adulteration should be configurable and automatically applied.
- SVT results and drug test results should appear on the same specimen record, linked, so that the complete validity and analytical picture is available to the certifying scientist before result authorisation.
- The reporting template for specimens with SVT flags should automatically route to the correct regulatory reporting pathway — substituted specimens to the MRO with the correct designation, not reported as negatives.
Failure 7 — Reporting Does Not Meet Medico-Legal or Regulatory Format Requirements
▸ The Problem
Toxicology laboratory reports are not generic result summaries. Depending on the testing context, they must meet specific format and content requirements: DOT Custody and Control Forms have a defined structure; MRO verification results must be reported in specific language; forensic toxicology reports for legal proceedings must include method descriptions, uncertainty statements, and certifying scientist credentials in specified formats; pain management monitoring reports may need to be structured for clinical integration into an EHR or practice management system.
A generic LIMS reporting module produces a standard laboratory report — patient name, test name, result, reference range, unit. This format satisfies almost none of the above requirements without extensive customisation. When the LIMS cannot produce the required report format natively, labs resort to exporting data and reformatting reports manually — a process that introduces transcription risk and is not sustainable at volume.
▸ Real-World Consequence
A forensic toxicology laboratory contracted to provide expert reports for criminal proceedings produces LIMS-generated reports in a generic format. Legal counsel for the defence challenges the chain of custody documentation embedded in the report and the absence of the certifying scientist's credentials and case number in the report header. The lab must manually produce a compliant report for each case — a two-hour exercise per case at volume.
How to Fix It
- Reporting templates in a toxicology LIMS must be configurable at the client, test type, and regulatory framework level. A single LIMS serving DOT and non-DOT clients, clinical and forensic clients, must be able to produce materially different report formats for different order types.
- For forensic reporting, the template must support inclusion of method summaries, instrumentation descriptions, quantitative uncertainty statements, certifying scientist credentials, and case-specific reference fields.
- For workplace drug testing, the LIMS should support electronic CCF reporting and direct interface with MRO software platforms where applicable.
- Report version control should be enforced — any amendment to a released report must be tracked, dated, authorised, and linked to the original report in the specimen record.
3. Generic vs. Toxicology-Configured LIMS: A Side-by-Side Comparison
The table below illustrates how the same workflow steps are handled differently by a generic LIMS deployment and a properly configured toxicology LIMS. The differences are not cosmetic — they reflect fundamentally different assumptions about what laboratory work looks like.
| Workflow Step | Generic LIMS | Toxicology-Configured LIMS |
|---|---|---|
| Sample Receipt | Logs receipt timestamp and accessioner. Chain of custody documentation not enforced. | Enforces e-signature at accessioning. Scanned CCF linked to specimen. Parent-aliquot relationship established at receipt. |
| Screen Positive Routing | Analyst manually flags and moves specimen to confirmation. No system-enforced routing. | LIMS auto-generates confirmation worklist on presumptive positive. Routing requires no manual intervention. |
| Cut-Off Application | Single global cut-off per analyte. Client or regulatory variation managed externally. | Client-level and order-level cut-off parameters. Version-controlled. Applied automatically to the correct batch. |
| Instrument Integration | Final quantitative result imported. Raw data file not linked to specimen record. | Result plus ion ratios, retention time, IS recovery imported. Raw file reference embedded in specimen record. |
| Specimen Validity | Processed as a separate test order. Manual cross-referencing with drug test results. | Integrated into specimen workflow. Automatic flag on threshold breach. Prevents drug result release until resolved. |
| QC Failure Response | QC failure noted. Manual decision on batch disposition. Variable documentation. | QC failure triggers automatic batch hold. Result release blocked. Override requires QA authorisation with documented rationale. |
| Report Generation | Generic result report. Manually reformatted for regulatory contexts. | Client-specific, regulatory-framework-specific templates. Forensic, DOT, and clinical report formats generated natively. |
4. What to Look for When Evaluating or Re-Configuring a Toxicology LIMS
Whether you are selecting a new LIMS for a toxicology facility or assessing whether your current system can be remediated, these are the questions and capabilities that will determine whether the system can genuinely serve the discipline.
During Vendor Evaluation
- Ask how many toxicology labs the vendor currently supports — and in which sub-disciplines. A vendor who supports five forensic toxicology labs has a fundamentally different understanding of the requirements than one who supports fifty clinical chemistry labs and claims the system can handle toxicology.
- Request a demonstration using a toxicology-specific scenario: a DOT urine screen batch with a presumptive positive that proceeds to GC-MS confirmation, with a substituted specimen in the same batch. Watch how the system handles the routing, the SVT flag, and the reporting. If the demo cannot be run without significant manual workarounds, the system is not toxicology-ready.
- Ask specifically about chain of custody enforcement — not tracking, enforcement. There is a difference. Ask the vendor to show you what happens in the system when a specimen is transferred from one analyst to another, and what the audit trail looks like for that transfer.
- Confirm that the system can produce the specific report formats you need — including any DOT-compliant reports, forensic expert report templates, or MRO interface files. Ask to see existing templates from comparable client sites, not new templates built for your demo.
During Re-Configuration of an Existing System
- Start with a gap assessment against the seven failure modes in this article. Document which gaps exist in your current configuration, which can be addressed through reconfiguration, and which would require fundamental architectural change that the platform may not support.
- Involve your certifying scientists and MRO interface contacts in the requirements gathering process. The people who sign out reports and manage regulatory submissions understand the output requirements better than anyone else in the lab.
- Prioritise chain of custody enforcement and SVT integration above all other configuration work. These are the gaps that carry the highest regulatory and legal exposure, and they are not recoverable through manual workarounds once a legal challenge arises.
- Any re-validation triggered by configuration changes must be scoped and resourced before changes are made, not after. Document the change impact assessment against your existing validation package before implementing any modification.
5. Frequently Asked Questions
Can a generic LIMS be configured to meet toxicology requirements, or does the lab need a purpose-built system?
It depends on the platform's flexibility and the depth of the toxicology requirements. Some enterprise LIMS platforms are genuinely configurable to the level that toxicology requires — but the configuration work is substantial, the scoping must be detailed, and the validation effort to support a toxicology-specific configuration is significant. Many labs have attempted to configure a generic platform for toxicology and found that certain requirements — particularly chain of custody enforcement at every transfer point and specimen validity integration — hit architectural limitations in the base platform. The question to ask a vendor is not 'can you configure this?' but 'show me where another toxicology lab is running this exact configuration in production.'
Our lab is DOT/SAMHSA regulated. What LIMS capabilities are non-negotiable?
For a lab performing DOT-regulated federal workplace drug testing under 49 CFR Part 40, non-negotiable LIMS capabilities include: chain of custody documentation at every specimen transfer point with electronic or wet-ink authentication; specimen validity testing integration with automatic flagging at substitution and adulteration thresholds; client-level cut-off management for DOT and non-DOT batches; QC enforcement meeting the specific calibrator and control requirements of Part 40; and report output that meets the CCF and MRO reporting requirements. Any LIMS evaluation for a DOT-regulated lab should be reviewed against the Part 40 requirements with legal or regulatory counsel.
How should a toxicology lab approach LIMS validation given the complexity of its workflow configuration?
Toxicology LIMS validation should be scoped with the full workflow complexity in mind from the beginning — not validated as a generic laboratory system and then extended. The URS must document every toxicology-specific requirement: chain of custody enforcement, two-stage routing, cut-off management, SVT integration, instrument integration scope, QC rules per method, and reporting formats. Each of these requirements should be tested during OQ and PQ with toxicology-specific test scripts — not generic laboratory scripts adapted from another discipline. For DOT-regulated testing, the validation package should also demonstrate that the system enforces Part 40 requirements in its configured state.
We currently manage chain of custody on paper forms alongside our LIMS. Is that acceptable?
Paper chain of custody forms are still required by regulation for some testing contexts, including DOT-regulated urine specimen collection at collection sites. However, a hybrid approach where the electronic LIMS record and the paper record are maintained in parallel but not formally linked creates a documentation gap that is increasingly difficult to defend. The stronger practice is to link the paper record to the electronic record at accessioning — scanned and embedded in the LIMS specimen record — so that the complete custody documentation is available in one place. If your current LIMS cannot support this, it is worth assessing whether the gap is configurable or architectural.
What questions should we ask when evaluating a LIMS vendor's toxicology experience?
Beyond standard product demonstrations, ask: How many toxicology laboratories are currently running your platform in production, and can you provide two or three references from facilities whose sub-discipline and regulatory context match ours? What is the process for configuring and validating a DOT-regulated testing workflow, and do you have a validated configuration we can review? Have any of your toxicology clients had their LIMS records reviewed in a legal proceeding, and what was the outcome? What is your vendor's process for keeping regulatory reference tables — cut-off values, SVT thresholds — current as guidance changes?
Final Thoughts
Toxicology is a discipline where the laboratory result is not just a clinical data point — it can determine employment, custody, freedom, or cause of death. The systems that generate and document those results carry a weight of accountability that generic LIMS implementations are not designed to bear.
The seven failures documented in this article are not edge cases. They are the standard experience of toxicology labs that have implemented LIMS platforms without accounting for the specific demands of the discipline. They are also correctable — either through reconfiguration of an existing system where the platform architecture allows it, or through migration to a platform with genuine toxicology depth.
The starting point in either case is the same: an honest assessment of where your current system meets the discipline's requirements, and where it is leaving you exposed. That assessment is worth doing before an audit, before a legal challenge, and before the next LIMS renewal conversation.
Continue Reading
- How to Prepare Your LIMS for an FDA Audit: A Practical Checklist
- Signs Your Legacy LIMS Is Costing You More Than You Think
- What Does a LIMS Validation Process Actually Look Like? A Step-by-Step Breakdown





 (1) (1).webp)



.webp)










