
How to Prepare Your LIMS for an FDA Audit: A Practical Checklist
Your LIMS is not just a data management tool. In the eyes of an FDA investigator, it is a regulated electronic records system — one that must comply with 21 CFR Part 11, uphold ALCOA+ data integrity principles, and demonstrate full traceability from sample receipt to final report. Any gap in these areas is a potential observation.
This checklist is written for Quality Assurance Managers, Lab Directors, and IT professionals at US-based laboratories. It covers the LIMS readiness areas that FDA investigators scrutinize most closely — so you can identify gaps before an auditor does.
Why This Matters in 2026
- FDA's emphasis on data integrity enforcement has intensified significantly over the past decade. Warning letters citing deficiencies in electronic records and audit trails have increased year over year.
- The FDA's Data Integrity and Compliance With Drug CGMP guidance (2018) and the accompanying Q&A document remain central references for regulated labs.
- Cloud-based and SaaS LIMS platforms have introduced new validation and access control considerations that traditional on-premise guidance does not fully address.
1. What FDA Investigators Look For in a LIMS
Before the checklist itself, it helps to understand how FDA investigators approach a LIMS during an inspection. Their primary concern is data integrity — the assurance that data are Attributable, Legible, Contemporaneous, Original, and Accurate (ALCOA), plus Complete, Consistent, Enduring, and Available (the '+' in ALCOA+).
During a LIMS review, investigators typically request:
- System validation documentation (IQ, OQ, PQ)
- Audit trail exports — including who changed what, when, and why
- User access control policies and privilege logs
- Electronic signature configuration records
- Change control history for system configurations
- Backup and disaster recovery procedures
- Computer system security and login records
Understanding these focal areas shapes how you approach every item on the list below.
2. The LIMS FDA Audit Readiness Checklist

Work through each section with your QA team. Any gap you identify should be addressed through your CAPA process and documented before your next inspection.
A. 21 CFR Part 11 Compliance
21 CFR Part 11 sets the requirements for electronic records and electronic signatures in FDA-regulated environments. Deficiencies here appear in warning letters more than any other LIMS-related area.
- Audit trail is enabled system-wide — All create, modify, and delete actions on regulated records are captured with user ID, timestamp, and reason for change.
- Audit trail records are protected — No user — including system administrators — can edit, delete, or disable audit trail entries.
- Audit trails are readily reviewable — Investigators should be able to filter and export audit trail data by date range, user, or record type without requiring IT intervention.
- Electronic signatures are compliant — Each e-signature is linked to one individual only; signing requires both a user ID and a password or biometric confirmation.
- Signature manifestations are complete — Every e-signature displays the signer's full printed name, date and time of signing, and the meaning of the signature (e.g., reviewed, approved, released).
- System access is role-based — Access privileges match job function; no user holds broader access than their role requires.
- Closed system controls are documented — Controls are formally documented per Part 11 requirements — standard for most laboratory environments.
B. Data Integrity and ALCOA+ Compliance

Data integrity is both a regulatory requirement and a laboratory culture issue. Your LIMS must technically support it — but your team must also practice it.
- All data entries are timestamped automatically — The system clock cannot be manipulated by standard users; any clock changes are logged.
- Original data is preserved — Corrections only overwrite when the original value is retained in the audit trail with a documented reason. Regulated data is never permanently deleted.
- Data is attributable to a specific individual — Every entry, modification, or review action links to a named, authenticated user — not a shared login.
- Shared logins are prohibited — The system configuration does not permit multiple individuals to share credentials. This is one of the most frequently cited FDA observations.
- Blank field controls are in place — The system prevents submission of incomplete records where data fields are required, reducing backdated entry risk.
- Metadata is captured and accessible — File metadata — including original creation date, modification history, and instrument data — is stored and accessible alongside results.
C. System Validation Documentation
FDA expects all software used in a GxP environment to be validated. Your LIMS validation package must be current, complete, and accessible within the timeframe an investigator expects.
- Validation Master Plan (VMP) is in place — Describes the validation strategy, scope, roles, and lifecycle approach for your LIMS.
- IQ/OQ/PQ documentation is complete and approved — Installation, Operational, and Performance Qualification documents are finalized, signed, and version-controlled.
- User Requirement Specifications (URS) are documented — Every validated function traces back to a defined user requirement.
- Validation is reconfirmed after system changes — Any update — including patches, configuration changes, and upgrades — triggers a documented change impact assessment and re-validation where appropriate.
- CSV documentation is stored and retrievable — Validation documents are available in paper or electronic form for review on request.
D. Access Control and User Management
- Access control policy is documented — A written SOP governs how user accounts are created, modified, and deactivated.
- User access is reviewed periodically — Formal access reviews — at minimum annually — are conducted and documented.
- Terminated or transferred employees are deactivated promptly — A defined process connects HR offboarding to LIMS account deactivation, with a clear SLA.
- Password policies meet minimum standards — Passwords expire periodically, meet complexity requirements, and cannot be reused.
- Failed login attempts trigger lockout — The system automatically locks accounts after a defined number of consecutive failed login attempts.
E. Change Control and Configuration Management
- All system changes go through formal change control — Configuration changes, SOP updates, and version upgrades are reviewed, approved, and documented before implementation.
- Change control records are complete — Each record includes the change description, risk assessment, impact on validation status, approvals, and implementation confirmation.
- A change history log for the LIMS is maintained — A complete chronological record of all changes since validation is available for review.
F. Backup, Disaster Recovery, and Business Continuity
- Automated backup schedules are in place and tested — Backup frequency matches the risk profile of your lab; restore tests are conducted and documented regularly.
- Backup integrity is verified — Periodic test restores confirm that data can be successfully recovered from backup media.
- Disaster recovery plan includes the LIMS — Your DR plan specifically addresses LIMS downtime scenarios, including manual backup procedures.
- Backup data is geographically separated — Primary and backup data are not stored on the same physical hardware or in the same building.
G. Instrument Integration and Raw Data Management
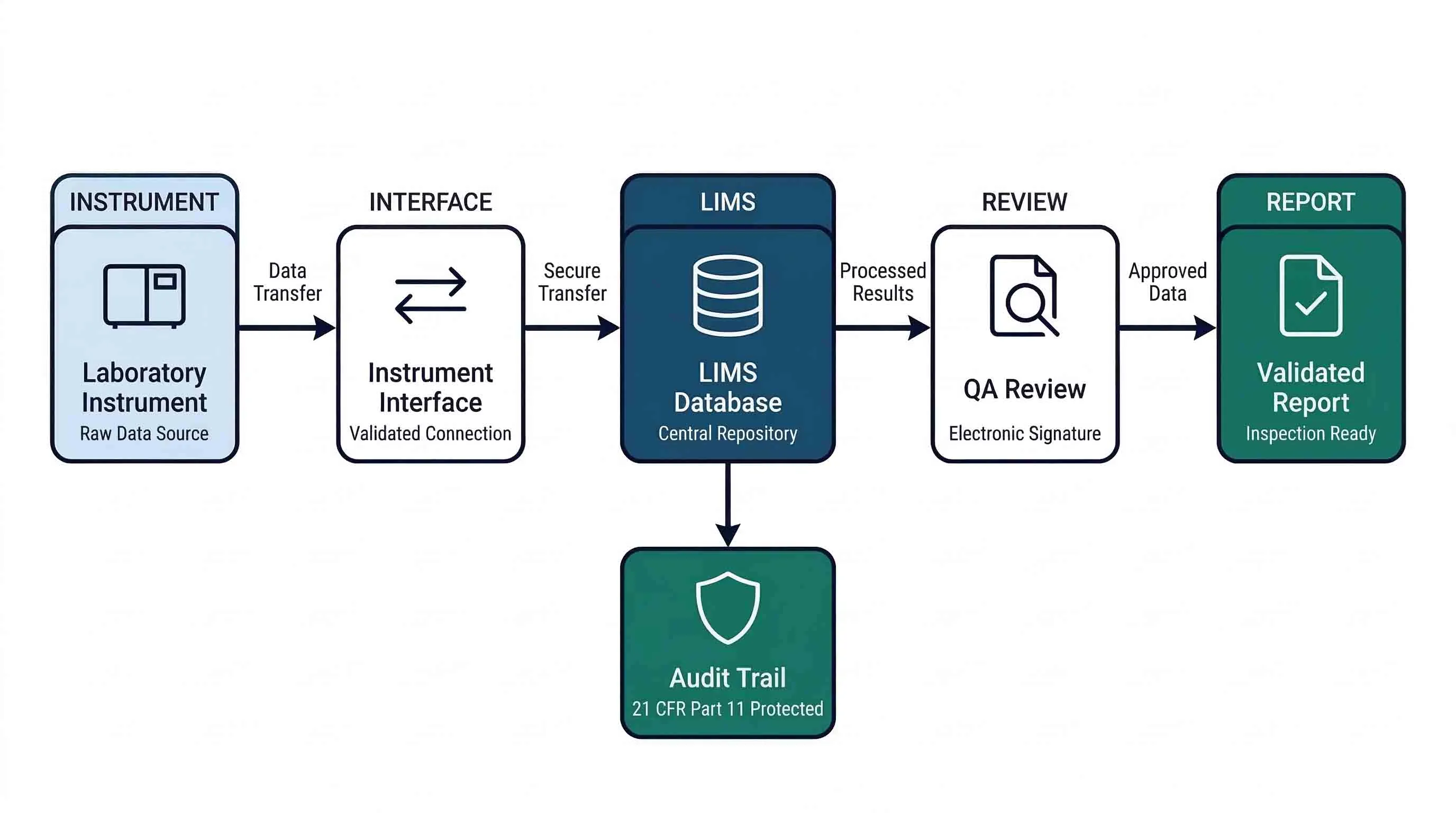
- Instrument interfaces are validated — Bidirectional interfaces between instruments and the LIMS are part of the validation scope and formally qualified.
- Raw data from instruments is captured and stored — The LIMS or an integrated system stores the original raw data file from each instrument — not just the final calculated result.
- Data transfer integrity is verified — Data transferred from instruments to the LIMS is confirmed complete and unaltered — ideally through a checksum or equivalent mechanism.
3. Common FDA Findings Related to LIMS — and How to Prevent Them
The following deficiencies appear repeatedly in publicly available FDA Warning Letters and 483 observations:

Common Audit Findings & Preventive Actions
| Common Finding | Preventive Action |
|---|---|
| Shared user accounts or generic logins | Enforce unique credentials for every user; disable group accounts in the LIMS configuration. |
| Audit trail disabled or incomplete | Verify audit trail coverage for all regulated records; include audit trail review in routine QA activities. |
| Lack of system validation documentation | Maintain a complete, accessible CSV package; include re-validation triggers in your change control SOP. |
| Administrator accounts used for routine work | Restrict admin-level access to IT/validation activities; auditors flag admin logins for daily work as a control gap. |
| Inability to retrieve historical data | Test data retrieval during periodic DR exercises; confirm legacy data from previous system versions remains accessible. |
| Electronic signatures not compliant | Review your e-signature workflow against Part 11 Subpart C; ensure all required manifestation fields are present. |
4. Building a Culture of Continuous Audit Readiness
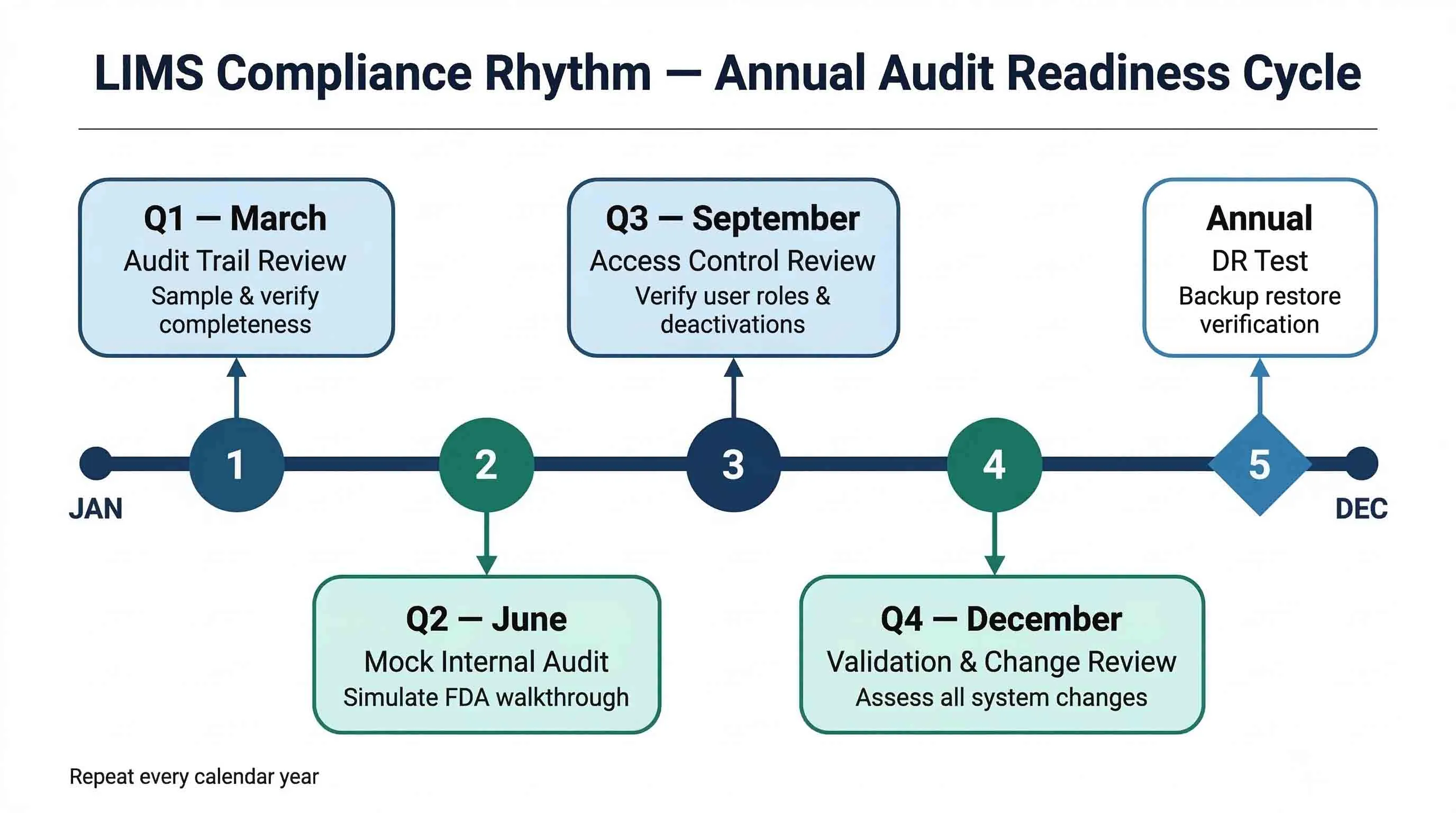
A checklist completed once before an inspection is not a compliance programme. FDA audit readiness requires an ongoing operational discipline, not a pre-inspection scramble. Labs that consistently fare well during inspections embed LIMS compliance activities into their routine quality management cycle:
- Quarterly audit trail reviews — QA staff periodically sample and review audit trail entries to verify completeness and accuracy.
- Annual access control reviews — verify that all active user accounts match current employees with appropriate role assignments.
- Change-triggered re-validation — every LIMS configuration change, upgrade, or patch is assessed for validation impact and documented accordingly.
- Mock audits — at minimum annually, conduct an internal walkthrough that simulates how an FDA investigator would review your LIMS, including requesting audit trail exports and validation documentation on the spot.
- Training records maintained in the LIMS — ensure training completions are documented and accessible, particularly for SOPs governing system use.
Key Regulatory References for US Laboratories
- 21 CFR Part 11 — Electronic Records; Electronic Signatures
- FDA Guidance for Industry: Data Integrity and Compliance With Drug CGMP (2018)
- FDA Guidance: Use of Electronic Records and Electronic Signatures in Clinical Investigations (2023 update)
- ICH Q10 — Pharmaceutical Quality System
- GAMP 5 (Second Edition, 2022) — Risk-Based Approach to GxP-Compliant Computerized Systems
5. Frequently Asked Questions
How often should we re-validate our LIMS?
There is no fixed regulatory interval for re-validation. The FDA expects validation to be maintained throughout the system lifecycle. In practice, every change — software updates, configuration modifications, infrastructure changes — should trigger a documented impact assessment. Where the assessment determines a change is significant, re-validation of the affected functions is required.
Does our LIMS vendor's validation package fulfil our Part 11 obligations?
No. While reputable LIMS vendors provide supporting documentation such as Installation Qualification protocols and system-level testing records, the regulatory responsibility for validating a computerised system in a GxP environment rests with the end-user organisation. You must produce your own validation package that reflects your specific configuration, workflows, and intended use.
What should we do if we identify a Part 11 gap right before an audit?
Document the gap immediately, initiate a CAPA, and assess the risk to data integrity. Do not attempt to correct records or alter system configurations without proper change control in place. Transparency with FDA investigators about identified gaps — alongside evidence of a structured remediation plan — is generally received more favourably than gaps that appear to have been overlooked or concealed.
Are cloud-based LIMS platforms subject to the same FDA requirements?Yes. The requirements under 21 CFR Part 11 and the FDA data integrity guidance apply regardless of whether your LIMS is hosted on-premise or in the cloud. For SaaS platforms, additional considerations apply — including vendor qualification, data sovereignty, subprocessor management, and the terms of your SaaS agreement as they relate to data ownership and audit trail access.
Final Thoughts
FDA audit readiness is not an event — it is an operational discipline. Laboratories that perform well during inspections are those that have made compliance a daily practice rather than a reactive exercise. Your LIMS sits at the centre of that practice: it should be configured so that compliant behaviour is the easiest behaviour, and it should generate the documentation that proves it.
The checklist above is a starting point. Every laboratory's regulatory context is different, and your inspection history, product types, and applicable regulations will shape your priorities. Use this as your framework, then layer in the specifics of your environment.
Author: Revol LIMS Team






 (1) (1).webp)



.webp)










