
The quality director of an international food and beverage manufacturer sits in a Friday afternoon compliance review meeting, staring at a spreadsheet that can no longer contain their reality.
Their company sells products in seven countries. Their laboratory is accredited by their national accreditation body. Their largest customer is FDA-regulated, demanding strict adherence to US standards. Their European distributor requires complete compliance with EU food safety regulations. Simultaneously, their Middle Eastern partner requires GCC Standardisation Organisation documentation, while their domestic regulatory body conducts its own exhaustive annual inspections.
Every single one of these regulators fundamentally wants the exact same thing: empirical evidence that the laboratory produces reliable, accurate, and traceable data. But each asks for it differently. They use different terminology, require distinct document formats, enforce different data retention periods, and mandate entirely different audit trail architectures.
The quality director sitting in this meeting is not managing one compliance programme. They are managing seven slightly different versions of the same compliance programme, simultaneously, utilising the same team, the same analytical instruments, and the exact same raw data.
"The multi-jurisdictional compliance burden is not a documentation problem. It is a fundamental infrastructure problem. And infrastructure problems require infrastructure solutions."
The laboratory that attempts to meet each of these requirements separately—utilising disconnected systems, manual transcriptions, or ad-hoc workarounds—ends up performing the exact same compliance work multiple times. This approach guarantees an increasing risk of critical compliance gaps between jurisdictions.
However, the laboratory that builds its global laboratory compliance infrastructure around the universal principles underpinning all major frameworks satisfies every regulator simultaneously from a single system. This is the strategic argument for modern technology architecture, and it is the argument this article will deconstruct.
- The universal compliance principles shared by every major global regulatory framework.
- The specific ways different regulatory frameworks diverge and where those divergences create the greatest compliance risk.
- The hidden cost of managing multi-jurisdictional compliance with manual or fragmented systems.
- What a laboratory information management system global infrastructure does at the architectural level to resolve multi-jurisdictional complexity.
- A practical framework for evaluating whether a laboratory's current infrastructure is genuinely multi-jurisdiction ready.
SECTION 1 — The Global Regulatory Landscape: More Unified Than It Appears
When viewed through the lens of daily operational paperwork, the global regulatory landscape appears as a fragmented, contradictory maze of requirements. However, when analysed at the architectural level, these global frameworks are expressions of a unified underlying philosophy.
Consider the major frameworks governing laboratory quality and multi-market food safety compliance today. In the United States, FDA 21 CFR Part 117 and the Food Safety Modernisation Act (FSMA) prioritise preventive controls and rigorous supply chain data. In India, FSSAI regulations enforce strict batch traceability and parameter-specific documentation.
Across the Atlantic, EU Regulation 178/2002 and the General Food Law framework emphasise granular, farm-to-fork traceability and rapid risk communication. Meanwhile, the Codex Alimentarius Commission sets the international baseline standards that underpin trade compliance across more than 180 countries, acting as the ultimate harmonising force in global food safety regulation.
In the laboratory itself, ISO 17025 governs testing and calibration accreditation globally, while GCC Standardisation Organisation requirements dictate specific market access rules across the Gulf. Overlaying all of this are the private supply chain schemes—FSSC 22000 and SQF—that international retail and foodservice sectors use to enforce their own quality mandates, alongside national standards like CFIA regulations in Canada.
We must stop viewing these frameworks as separate, isolated silos. They are merely different regional dialects of the same fundamental language. Food safety and laboratory quality are universally built on data integrity, traceability, documentation completeness, and evidence-based decision-making.
Every single one of these frameworks shares five universal compliance principles: complete traceability of materials and products, absolute integrity and immutability of quality data, complete documentation of testing processes and results, verified evidence of instrument calibration, and rapid retrievability of records for audit.
These five principles are the foundation of global compliance. A laboratory compliance infrastructure built to natively satisfy these five principles simultaneously satisfies the underlying requirement of every major global framework. The challenge is no longer understanding the regulations; the challenge is deploying the architecture to meet them seamlessly.
How many different regulatory frameworks are your laboratory currently required to satisfy simultaneously? Do you have a single system of record for quality data that satisfies all of them, or do you maintain separate documentation for different markets? If a single auditor from any one of your regulatory bodies requested your complete batch traceability records tomorrow, how long would it take to produce them?
SECTION 2 — Where the Frameworks Diverge: The Seven Compliance Fault Lines
While the underlying philosophy of global regulation is unified, the operational execution is where the danger lies. The specific areas where major global regulatory frameworks diverge represent compliance fault lines. For laboratories operating across multiple jurisdictions, these fault lines are where data gaps, human errors, and audit failures incubate.

Fault Line 1 — Electronic Record and Electronic Signature Requirements
The transition from paper to digital records is fraught with jurisdictional divergence. FDA 21 CFR Part 11 defines electronic records and electronic signatures through highly specific, rigid technical requirements regarding password ageing, biometric linkages, and user authentication protocols.
If a laboratory wishes to use electronic systems for FDA-regulated records, it must meet these exact specifications. However, the European equivalent, Annex 11 of EudraLex, approaches the same requirement with different technical specifications, focusing more broadly on risk management and system validation.
For a laboratory serving both markets, satisfying both simultaneously requires a system that defaults to the strictest technical interpretation of both frameworks, ensuring user access and signature workflows meet the uncompromising biometric and password standards of the FDA while satisfying the risk-based validation requirements of the EU.
Fault Line 2 — Data Retention Periods
Data retention is rarely harmonised globally. FDA regulations mandate highly specific retention periods based on product shelf life and record category. Meanwhile, FSSAI requirements in India, EU food law, and ISO 17025 all dictate varying timelines for retaining calibration logs, test results, and raw data.
The root cause of failure occurs when a laboratory sets a blanket data retention policy based on its domestic framework, entirely missing the fact that a foreign framework it is subject to requires records to be held two years longer.
In a fragmented data ecosystem—where CoAs live in a shared drive and instrument logs live in physical binders—implementing, tracking, and verifying a comprehensive, multi-jurisdictional retention policy is practically impossible.
Fault Line 3 — Traceability Depth and Scope Requirements
Traceability means different things to different regulators. FDA FSMA heavily enforces a "one-step-forward, one-step-back" traceability minimum. Conversely, the EU General Food Law, bolstered by more recent digital traceability initiatives, pushes toward a much more granular, unbroken farm-to-fork continuum.
Furthermore, Codex Alimentarius compliance principles on traceability underpin the baseline requirements of multiple national frameworks simultaneously.
For a manufacturer exporting globally, this divergence creates a strategic imperative: the traceability depth you build for your most demanding market effectively satisfies all your less demanding markets. However, if your system cannot aggregate data at that granular EU level, your market access remains structurally limited to jurisdictions with lower traceability thresholds.
Fault Line 4 — Certificate of Analysis Format and Content Requirements
The Certificate of Analysis (CoA) is the passport of international trade, yet its required format varies wildly. FDA-regulated customers expect specific formatting regarding methodology and limits, EU customers require adherence to European parameter thresholds, and Indian FSSAI-regulated products must display specific regulatory identifiers.
"When producing market-specific CoAs requires manual reformatting of the same underlying data, the laboratory is actively injecting data integrity risk into its most critical commercial documents."
In a manual CoA system, analysts must manually reformat the same underlying test data for each distinct market. This creates a severe data integrity risk. When the same batch result appears on three different CoA formats generated manually by three different analysts on different days, the probability of transcription errors leading to regulatory rejection approaches certainty.
Fault Line 5 — Audit Trail Coverage and Depth
The expectation of what an audit trail must capture differs across borders. 21 CFR Part 11 requires strict, computer-generated, timestamped audit trails that independently record the date and time of operator entries and actions. ISO 17025 clause 8.4.2 defines the required scope of quality records differently, focusing on technical retrievability.
Today, ALCOA+ compliance principles influence audit trail expectations across almost all frameworks, even where not explicitly written into law.
Complete ALCOA+ compliance requires data to be Attributable, Legible, Contemporaneous, Original, and Accurate, plus Complete, Consistent, Enduring, and Available. Achieving this comprehensive standard in a manual, paper-based, or fragmented software system is effectively impossible at the scale required by modern global supply chains.
Fault Line 6 — Proficiency Testing and External Quality Assurance Requirements
External validation is a universal requirement, but its execution is hyper-localised. Different accreditation frameworks specify vastly different expectations for proficiency testing (PT) participation.
ISO 17025 lays out baseline requirements for accredited laboratories, but regulators in different markets view PT evidence differently during an inspection. Some markets mandate participation in very specific, state-sponsored PT schemes for certain test categories.
Managing PT records across multiple schemes becomes a severe administrative burden when different customers and regulators require distinct evidence of participation. If a laboratory cannot immediately produce the correct, market-specific PT evidence for an analytical method during a multi-jurisdictional audit, the validity of every result generated by that method is questioned.
Fault Line 7 — Incident Reporting and Corrective Action Documentation
When a process fails, the frameworks diverge on how the failure must be documented. The Corrective and Preventive Action (CAPA) documentation requirements under FDA frameworks differ structurally from the non-conformance management requirements under ISO 17025, which in turn differ from the corrective action expectations in FSSC 22000 audits.
Corrective action documentation remains one of the most commonly cited areas of audit observation globally. Fragmented quality systems make adequate CAPA documentation structurally difficult to achieve because the original failure (recorded in one system) cannot be seamlessly linked to the root cause analysis and the final corrective action verification (recorded in separate systems).
- Your laboratory maintains different documentation systems or distinct software tools for different target markets.
- Your CoA format changes manually depending on which customer or market the product is destined for.
- Your audit trail coverage is inconsistent, capturing deep data for certain regulatory contexts while ignoring others.
- Your data retention policy was last reviewed more than two years ago and was written with only a single regulatory framework in mind.
SECTION 3 — The Hidden Architecture Problem: Why Multi-Jurisdictional Compliance Fails
When sophisticated laboratories with capable, highly trained quality teams fail multi-jurisdictional audits, the root cause is rarely human negligence. The failure is almost always architectural.
There is a fundamental, unresolvable incompatibility between fragmented, manual data systems and the rigorous demands of simultaneous multi-jurisdictional compliance. This failure manifests across four distinct failure modes.

Failure Mode 1 — The System of Record Problem
In many global laboratories, quality data lives in multiple, disconnected silos. Instruments write raw data to local proprietary software. Analysts manually transcribe those results to shared spreadsheets. Final CoAs are generated in basic word processor templates. Calibration records live in a separate, physical folder structure, while batch records reside in a corporate ERP system that does not communicate with the laboratory.
There is no single authoritative system of record. In a multi-jurisdictional compliance context, this fragmentation is catastrophic. Different regulators may be shown slightly different data for the exact same analytical event, depending entirely on which isolated system was consulted when the compliance records were manually compiled. This lack of a single source of truth destroys regulatory trust instantly.
Failure Mode 2 — The Translation Tax
Every single time a laboratory must produce compliance documentation for a different regulatory framework using the same underlying data, it pays a translation tax.
This tax is paid in highly skilled analyst hours spent reformatting records, reorganizing data structures, relabeling parameters to match different frameworks' terminology, and recreating documents that already exist in a different format.
This translation tax is largely invisible in daily operations, but it is enormous in aggregate. If a mid-sized laboratory serves four different regulatory markets and produces distinct monthly compliance reports for each one, this translation tax easily consumes thousands of analyst hours annually. This is time that should be spent on complex quality science and continuous improvement, entirely wasted on administrative data translation.
Failure Mode 3 — The Gap Accumulation Problem
Multi-jurisdictional compliance failures almost never happen all at once in a spectacular collapse. They accumulate gradually, silently. Small gaps between different systems create small inconsistencies that are never individually significant enough to trigger a formal corrective action, but collectively represent a material compliance crisis.
A calibration record maintained in one system does not perfectly match the instrument serial number recorded in the testing system. A batch number format in the manufacturing system utilizes a hyphen, while the CoA in the quality system utilizes a slash. A retention period applied in a physical document archive does not match the stated retention policy in the laboratory's quality manual.
Each gap is incredibly small. Together, when viewed by an international auditor cross-referencing your systems, they constitute a systemic audit finding.
Failure Mode 4 — The Knowledge Concentration Risk
In laboratories that manage complex multi-jurisdictional compliance manually, the knowledge of how to navigate between different frameworks' requirements typically resides in the minds of one or two senior personnel.
It is the veteran quality manager who knows exactly which specific form goes to the FDA, which distinct data format satisfies the EU, and which calibration proof the domestic regulator prefers.
When those people leave the organization, that critical compliance knowledge leaves with them. The laboratory abruptly discovers how dangerously dependent it was on individual expertise rather than institutional system capability. This realization almost always occurs precisely at the moment the laboratory can least afford it: during a recruitment gap or a high-stakes, unannounced regulatory inspection.
If your most experienced quality manager left the company tomorrow, exactly how long would it take your remaining team to produce a complete, error-free compliance package for your most demanding regulatory framework? Is your multi-jurisdictional compliance capability a hardcoded property of your systems, or merely a fragile property of specific individuals?
SECTION 4 — The Real Cost of Multi-Jurisdictional Compliance Without Adequate Infrastructure
Operating a global laboratory without adequate technology infrastructure is an active, highly expensive financial decision. The costs extend far beyond the laboratory's operational budget, directly impacting corporate revenue and strategic growth. We must quantify these costs honestly across five specific categories.
Cost Category 1 — The Translation Tax Quantified
Let us calculate the true cost of the translation tax for a mid-size laboratory serving four distinct regulatory markets. If four analysts spend just ten hours a week manually reformatting data, generating market-specific CoAs, and compiling distinct compliance reports, that is 2,000 labour hours consumed annually.
Add the senior quality team hours consumed by the mandatory review and approval of these multiple, market-specific documentation variants. Expressed as a labour cost, this runs into tens of thousands of dollars annually. Expressed as an opportunity cost, the calculation is much higher: this is 2,000 hours of high-level quality science, process optimisation, and system development work that is simply not happening.
Cost Category 2 — The Audit Failure Cost
The financial and operational consequences of a multi-jurisdictional compliance failure are cascading. A critical finding is never isolated to a single market.
"A 483 observation from the FDA does not stay local. It becomes immediately visible to global customers who conduct their own rigorous supplier qualification reviews."
If an ISO 17025 non-conformance is identified during a NABL or UKAS reassessment visit, it has immediate implications for the laboratory's overarching accreditation status. This, in turn, suspends the commercial validity of the results it has produced for customers in multiple markets simultaneously. The cost involves hiring external remediation consultants, shutting down production lines, conducting massive product recalls, and enduring the severe reputational damage that causes global partners to seek alternative suppliers.
Cost Category 3 — The Market Access Cost
The inability to credibly demonstrate compliance with a new market's regulatory requirements actively blocks revenue. When a company wishes to export to a new, highly regulated jurisdiction, but the laboratory's infrastructure cannot reliably produce the specific digital documentation that the market requires, market entry is halted.
This cost is not just the compliance consultant fees required to build temporary workarounds. It is the agonising delay in market entry, the millions in potential revenue foregone during that delay, and the massive competitive advantage freely handed to a rival manufacturer who already has the multi-jurisdictional infrastructure in place to move immediately.
Cost Category 4 — The Operational Inefficiency Cost
Running multiple parallel documentation systems carries a massive operational inefficiency cost. A laboratory without a unified infrastructure must pay software licensing fees for various disconnected systems that serve separate compliance purposes.
There are physical and digital storage costs for maintaining documentation in multiple formats across multiple retention periods. Furthermore, there is the heavy IT support cost of maintaining these siloed systems, coupled with the daily coordination cost of ensuring they somehow remain consistent with each other over time as both the software and the global regulatory requirements continue to evolve independently.
Cost Category 5 — The Strategic Cost
The most severe cost of inadequate infrastructure is the strategic cost that never appears on a laboratory budget spreadsheet.
It is the cost of organisational paralysis. A laboratory that is currently operating at maximum administrative capacity just to maintain its current multi-jurisdictional obligations cannot pursue new global markets, accept new international customers, or support new product categories. The laboratory that has outgrown its compliance infrastructure is not just paying a compliance cost; it is paying a terminal growth cost.
SECTION 5 — The Universal Architecture: How LIMS Infrastructure Resolves Multi-Jurisdictional Compliance
Resolving this complex, multi-layered burden requires shifting from a procedural mindset to an architectural one. Modern Laboratory Information Management Systems (LIMS) resolve the multi-jurisdictional compliance challenge by hardcoding the five universal compliance principles directly into the laboratory's digital infrastructure.
When you build an ISO 17025 LIMS architecture correctly, the system natively satisfies multiple regulatory frameworks simultaneously from a single, unified dataset.
Principle 1 — Complete Traceability
A modern, multi-jurisdictional LIMS creates and maintains complete forward and backward traceability chains entirely automatically. From the moment an incoming raw material or sample lot is logged, the system tracks every single processing step, testing event, instrument utilised, analyst responsible, result generated, CoA issued, and dispatch record created.
This single, automated traceability architecture natively satisfies the strict traceability requirements of FDA FSMA, EU General Food Law, FSSAI Schedule 4, Codex Alimentarius principles, and ISO 17025 simultaneously, without any additional documentation effort required for different markets. Because the data is complete and automatically linked at the exact time of creation, producing traceability evidence for any specific regulatory framework is simply a matter of selecting the appropriate report format. The underlying data is identical.
Principle 2 — Data Integrity and Immutability
A proper laboratory information management system's global architecture enforces data integrity at the deepest system level. It generates electronic records that simply cannot be altered without creating a permanent, indelible audit trail entry.
By utilising direct instrument data transfer, the system entirely eliminates manual transcription errors. Strict user access controls ensure only authorised personnel can create or modify regulated records, while timestamp integrity ensures records can never be backdated. This architecture simultaneously satisfies 21 CFR Part 11 electronic records requirements, EU Annex 11 requirements, ISO 17025 clause 7.11 data integrity standards, and overarching ALCOA+ compliance principles across all frameworks from a single implementation.
Principle 3 — Documentation Completeness
The most common cause of audit findings globally is missing information. LIMS enforces documentation completeness through rigid, system-level workflow controls.
Sample login workflows cannot be completed without all required fields populated. Test result workflows demand explicit analyst review and supervisor electronic approval before any result can be reported. Automated CoA generation is physically blocked by the system until all required parameters possess verified results and all results have been reviewed against the correct market specifications. These workflow controls prevent documentation gaps not by adding more human checks, but by making incomplete documentation architecturally impossible within the system.
Principle 4 — Instrument Calibration and Maintenance Evidence
Global regulators demand proof that instruments were performing correctly at the exact moment a test was run. A modern LIMS maintains complete, integrated instrument records: precise calibration schedules, calibration results, and maintenance histories.
Crucially, it automates the linkage between an instrument's calibration status and the specific test results produced on that instrument. If a balance is out of calibration, the LIMS blocks the analyst from using it. This automated linkage satisfies the instrument traceability requirements of ISO 17025, the equipment qualification requirements of FDA GMP frameworks, and instrument validation requirements across multiple other frameworks simultaneously.
Principle 5 — Rapid Record Retrievability
When all quality data lives in a single, authoritative system of record with complete relational linkages between related files, producing a complete compliance package for any regulatory framework transforms from a multi-day panic into a matter of minutes.
"For a laboratory subject to unannounced international inspections, rapid retrievability provides the ultimate operational confidence: complete, audit-ready documentation can be produced for any market at any moment."
This rapid retrievability fundamentally changes the economics of expanding into new regulatory markets. If your global infrastructure already captures all the underlying data securely and comprehensively, the incremental cost of producing new compliance documentation for a newly entered framework is virtually zero.
Laboratories operating with proper LIMS infrastructure gain a massive strategic advantage, known as the multi-jurisdiction dividend. This is the ability to enter new, highly regulated global markets without suffering proportional increases in compliance overhead. It is the capacity to onboard customers from entirely new regulatory environments without purchasing or building new documentation systems. It is the power to demonstrate absolute compliance readiness to any auditor, from any framework, at any given time.
SECTION 6 — Building a LIMS Business Case for Multi-Jurisdictional Compliance: What Leadership Needs to See
Quality professionals intuitively understand the absolute necessity of modern LIMS infrastructure. However, securing the budget requires convincing financial and executive leadership, who frequently mischaracterise LIMS as a standard IT project rather than a critical compliance and market-access investment.
Building a successful business case for multi-jurisdictional LIMS requires speaking the language of risk and revenue.
Component 1 — The Current State Cost Map
Begin by guiding your leadership through a highly specific map of the full cost of your current multi-jurisdictional compliance approach. Quantify the "translation tax" discussed earlier. Calculate the heavy system maintenance overhead of running multiple disconnected spreadsheets and software silos. Tally the massive labour costs dedicated to manual audit preparation.
Most importantly, articulate the knowledge concentration risk. The final financial figure that emerges from this honest exercise forms the absolute foundation of the ROI argument.
Component 2 — The Market Access Argument
Never frame a LIMS investment purely as a compliance cost. Frame it as a market access enabler.
Every new regulated global market the laboratory can credibly serve represents massive incremental revenue. Conversely, the laboratory's current manual compliance infrastructure is actively preventing the organisation from accessing these markets. Explain to leadership that the modern LIMS infrastructure investment directly unlocks this revenue.
Component 3 — The Risk Quantification
Executive leadership understands risk when it is properly quantified. Provide a stark framework detailing the compliance risk the current infrastructure carries.
Estimate the probability of a significant, multi-market audit finding occurring within the next three years based on your current manual gap accumulation. Detail the devastating financial and commercial consequences of such a finding. When this quantified risk is expressed as an expected annual cost, it routinely dwarfs the annual cost of the LIMS infrastructure.
Component 4 — The Competitive Positioning Argument
Finally, frame the investment in terms of the harsh competitive landscape. Highlight the advanced compliance capabilities, real-time reporting, and absolute data integrity that your laboratory can demonstrate to global customers and regulators that competitors cannot. The argument is clear: the market is moving to digital compliance; those left on paper will simply not be able to compete.
SECTION 7 — Evaluating LIMS for Multi-Jurisdictional Compliance: The Questions That Actually Matter
When evaluating a laboratory information management system for global deployment, generic software questions are insufficient. Quality professionals must ask the specific, probing questions that distinguish systems built for genuine multi-jurisdictional complexity.
- Evaluating Electronic Records: Do not ask if the system has electronic signatures. Ask exactly how the system's electronic record and signature implementation simultaneously satisfies the rigid biometric and password ageing requirements of 21 CFR Part 11 alongside the risk-based validation requirements of EU Annex 11.
- Evaluating Audit Trails: Do not ask if the system has an audit log. Ask how the system evaluates audit trail completeness, specifically against the nine pillars of ALCOA+ principles.
- Evaluating Configurability: Ask how the system handles report and CoA configurability for different market documentation requirements without altering the underlying raw data.
- Evaluating Retention: Ask how the system enforces data retention policy implementation across different record categories based on intersecting regulatory rules.
"Do not ask if the system is compliant. Ask how rapidly and reliably the system vendor deploys architectural updates when global regulatory frameworks inevitably evolve."
- Evaluating Instrument Integration: Evaluate the instrument interface capabilities. Does the system rely on file parsing, or does it offer direct, bi-directional data transfer that entirely eliminates transcription risk?
- Evaluating Regulatory Adaptability: Finally, evaluate the vendor's regulatory update process. A LIMS is a long-term partnership; the vendor must be a regulatory expert, not just a software developer.
SECTION 8 — A Practical Framework for Multi-Jurisdictional Compliance Readiness Assessment
Before beginning a technology procurement process, a laboratory must rigorously evaluate its current state. Quality professionals can use this practical four-dimensional assessment to evaluate their laboratory's genuine multi-jurisdictional compliance readiness.
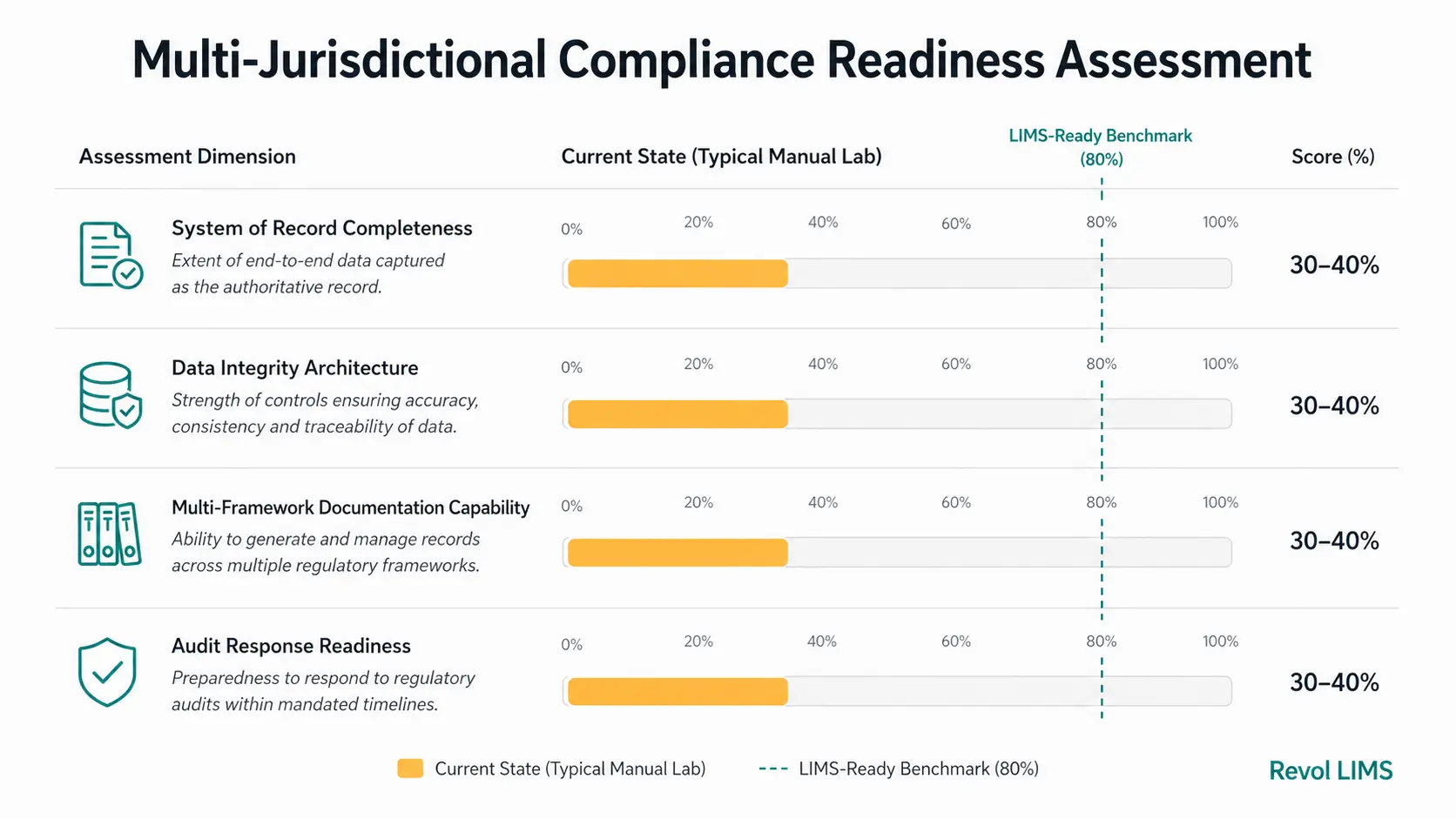
Assessment Dimension 1: System of Record Completeness
- Does a single, unified software system capture all regulated quality data (instruments, training, testing, calibration), or is critical data spread across multiple disconnected systems and physical logs?
- Are all raw data files securely linked to the final reported results?
- Can you track a sample's complete lifecycle without leaving the primary software interface?
Assessment Dimension 2: Data Integrity Architecture
- Does the system enforce data integrity automatically at the architectural level, or does it rely heavily on procedural controls and human compliance?
- Are audit trails computer-generated, indelible, and independently timestamped?
- Is it physically impossible for an analyst to delete a failed test result without authorisation and a mandatory reason code?
Assessment Dimension 3: Multi-Framework Documentation Capability
- Can the system instantly produce fully compliant documentation (CoAs, batch records, compliance summaries) for all your required global regulatory frameworks from a single, centralised data set?
- Does producing documentation for a new market require manual recalculation or data transcription?
Assessment Dimension 4: Audit Response Readiness
- If an auditor arrived unannounced, could the laboratory produce a complete, flawless compliance package encompassing full traceability, calibration evidence, and PT participation for any required regulatory framework within one hour?
- Does audit preparation require mandatory weekend overtime for the quality team?
Frequently Asked Questions
Our laboratory is already ISO 17025 accredited — does that mean we are effectively compliant with FDA and other major frameworks as well?
No. While ISO 17025 demonstrates exceptional technical competence and is a foundational achievement, it does not automatically satisfy the specific legal and procedural mandates of the FDA, FSSAI, or EU regulators. ISO 17025 focuses heavily on laboratory processes and measurement uncertainty, whereas frameworks like FDA 21 CFR Part 11 focus rigidly on electronic record security. A modern LIMS bridges this gap by applying ISO 17025 rigour to FDA and EU data structures. Our laboratory is already ISO 17025 accredited, demonstrating our commitment to these global standards.
We operate in three different countries with three different ERP systems — can a LIMS integrate with all of them, or does it create another siloed system?
A truly global, enterprise-grade LIMS acts as the centralised scientific hub and is designed to integrate seamlessly via robust APIs with multiple disparate ERP systems across different geographies. It acts as the ultimate system of record for quality data, eliminating silos rather than creating them.
How does a LIMS handle regulatory framework updates — when the FDA or another body issues new guidance, how does the system adapt?
Top-tier global LIMS providers operate dedicated regulatory compliance teams. When major frameworks evolve—such as updates to ALCOA+ guidance or new EU traceability mandates—the vendor issues validated system updates. Because a modern LIMS is highly configurable, quality teams can also rapidly adjust internal workflows, specifications, and report templates to meet new guidance without requiring hardcoded software changes.
Our laboratory produces results that are used by customers in twelve different countries — do we need to configure the LIMS differently for each market, or can one configuration serve all markets?
You configure the data capture process once, utilising the strictest common denominator of all your target markets to ensure maximum data integrity and traceability depth. You then configure the reporting outputs (like CoAs and compliance summaries) in twelve different ways. The brilliance of a properly implemented LIMS is that the rigorous, unified underlying dataset seamlessly populates any necessary regional report format instantly.
FINAL THOUGHTS
The laboratories and manufacturers managing multi-jurisdictional compliance today are navigating a genuinely complex and highly consequential challenge. The global regulatory frameworks they operate under were never designed in coordination with each other.
However, the fact that these diverse frameworks share common, underlying principles is not a regulatory design decision. It is a reflection of a universal truth: quality and safety in any industry ultimately depend on the same things. They depend on reliable, immutable data; complete, contemporaneous documentation; and the absolute ability to reconstruct exactly what happened to any material, product, or sample at any point in its journey.
"The laboratory that builds its infrastructure around universal truths rather than specific documentation requirements will find that global compliance becomes a strategic capability rather than a localised burden."
If your laboratory builds its technology infrastructure exclusively around the specific, localised documentation requirements of a single framework, you will constantly chase compliance. But if you architect your infrastructure around these universal truths of data integrity and traceability, multi-jurisdictional compliance becomes a natural byproduct of your daily operations.
We invite you to look closely at your laboratory today. Evaluate, with absolute honesty, whether your current infrastructure is genuinely built for the relentless, multi-market compliance landscape you operate in today. Compliance is no longer a paperwork exercise; it is the ultimate test of your infrastructure's integrity.





 (1) (1).webp)



.webp)










